Paul G Hynes, Kathleen Kelly
Cell and Cancer Biology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
| Date of Submission | 04-Oct-2011 |
| Date of Acceptance | 10-Nov-2011 |
| Date of Web Publication | 12-Mar-2012 |
Correspondence Address:
Kathleen Kelly
Cell and Cancer Biology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD
USA
Source of Support: None, Conflict of Interest: None
DOI: 10.4103/1477-3163.93701
Abstract
Advanced prostate cancers are treated with androgen deprivation therapy, which usually leads to a rapid and significant reduction in tumor burden but subsequent development of castration-resistant and metastatic disease almost always occurs. The source of tumor heterogeneity and the accompanying mechanisms leading to treatment resistance are major areas of prostate cancer research. Although our understanding of tumor heterogeneity is evolving, the functional isolation of tumor propagating populations, also known as cancer stem cells (CSCs), is fundamental to the identification and molecular characterization of castration-resistant prostate cancer cells. Of clinical importance, knowledge of prostate CSCs has implications for design of next generation-targeted therapies aimed at both eradicating primary tumor mass and preventing castration-resistant disease. The inability to routinely transplant fractionated primary human prostate tumors has prevented progress in analyzing the source of heterogeneous and treatment-resistant populations in prostate cancer. Here, we briefly overview the mechanisms of castration resistance, including the hypothesis for the existence of androgen-independent prostate CSCs. Finally, we discuss the interpretation of preclinical models and their utility for characterizing prostate CSCs in androgen-replete and androgen-deprived conditions.
Keywords: Animal models, cancer stem cells, castration resistance, prostate cancer
How to cite this article:
Hynes PG, Kelly K. Prostate cancer stem cells: The case for model systems. J Carcinog 2012;11:6
How to cite this URL:
Hynes PG, Kelly K. Prostate cancer stem cells: The case for model systems. J Carcinog [serial online] 2012 [cited 2021 Oct 14];11:6. Available from: https://carcinogenesis.com/text.asp?2012/11/1/6/93701
Introduction
Prostate cancer is the second leading cause of cancer death in men, and is predicted to account for the highest number, approximately 30%, of newly diagnosed male cancer cases in 2011. [1] Prostate cancer-related deaths occur predominantly as a result of metastatic disease that is resistant to androgen deprivation therapy (ADT), usually referred to as castration resistance. Major clinical challenges exist for identifying aggressive prostate cancers that are likely to metastasize and for targeting treatment to non-androgen receptor (AR) pathways. Understanding the mechanisms underlying castration-resistant progression is an important area of basic research in prostate tumor biology. Fundamental to investigating castration-resistant mechanisms is the definition of cell populations capable of propagating prostate tumor growth and metastasis in androgen-replete and -deprived conditions.
The ability of subsets of tumor cells upon transplantation to self-renew and recapitulate the heterogeneity of the original tumor is at present the major criteria for defining cancer stem cells (CSCs). Tumor cell subpopulations with such capacity, also called tumor-propagating cells or tumor-initiating cells, have been identified in a variety of malignancies and vary greatly in their frequencies. [2],[3] In some cancers, nearly every cell has tumor-propagating ability. [4] In addition, the quantification of CSCs is not an absolute but, instead, depends on the specifications of the assay. Nevertheless, the quantification and molecular characterizations of CSC populations from primary tumors have been useful in gauging tumor aggressiveness and heterogeneity. [5],[6]
The search for CSCs within human prostate cancer is ongoing. Currently, a major obstacle in the field is the inability to routinely grow freshly dissociated human prostate cells in mouse transplant recipients. Here, we discuss the utility of identifying CSCs in prostate cancers, review the current models for prostate cancer cell of origin and the relationship to CSCs and pose what we believe to be the significant value of preclinical models to begin addressing the heterogeneity of prostate tumors and the molecular characterization of tumor propagating subpopulations.
Prostate Cell Lineages and Regenerative Potential
Human and mouse prostates have a glandular structure composed of three epithelial cell types: basal, luminal and neuroendocrine cells, embedded within a fibromuscular matrix. [7] Basal cells rest on basement membranes and are characterized by their expression of markers such as cytokeratin 5 (CK5), CK14 and p63. In addition, they express heterogeneous, low levels of AR. Columnar luminal epithelial cells reside above the basal cells and express CK8, CK18, Nkx3.1, high levels of AR and, in the case of human cells, prostate-specific antigen (PSA). Luminal cells are androgen dependent and, on castration, most luminal cells apoptose, although in the mouse, a single layer of attenuated, cuboidal luminal cells survives above the basal layer. Finally, rare androgen-independent neuroendocrine cells are marked by the expression of synaptophysin, chromogranin A or beta-3 tubulin.
The existence of epithelial stem cells in the adult prostate is implied by the ability of the prostate to undergo multiple rounds of castration-induced regression followed by full regeneration upon restoration of androgen. [8],[9] Following cell surface labeling and fluorescence-activated cell sorting (FACS), cells that fractionate with the basal compartment have been shown to self-renew and are capable of generating basal and luminal cells. [10],[11] Fractionation and transplantation assays have demonstrated that a single Lin – Sca-1 + CD133 + CD44 + CD117 + cell, within the basal fraction, can give rise to luminal, basal and neuroendocrine cell types in the mouse prostate. [12] In addition, there also exist in the mouse castration-resistant NKX3.1 + luminal cells, termed CARNs, with bipotential regenerative activity. [13]
Prostate Cancer Pathology
Prostate adenocarcinoma is believed to develop from early precursor lesions known as prostatic intraepithelial neoplasia (PIN). [14] Prostate glands undergo numerous changes during the course of progression, including the loss of basal cells and the hyperproliferation and morphological transformation of luminal cells. [15] These cellular changes result in the majority of prostate cancers having a pronounced luminal phenotype and being classified histologically as acinar adenocarcinomas. A noteworthy feature of prostate cancer is the rare occurrence of other histopathological subtypes. [16],[17] Possible interpretations of this observation are that: (1) a predominant cell compartment in the prostate is susceptible to transformation and/or (2) transformation of various cells within a related lineage leads to the expression of a similar differentiated phenotype. One characteristic of adenocarcinoma is a lack of basal cells. By contrast, the accepted precursor lesion of adenocarcinoma, high-grade PIN (HGPIN), retains some evidence of basal cells. Thus, HGPIN lesions are useful entities defined within a tumor progression paradigm to evaluate cell populations harboring mutations. Importantly, a variety of cancer-associated markers are expressed in luminal but not basal phenotyped cells of HGPIN. [18],[19],[20] The most direct interpretation of these data is that transforming mutations do not occur in histologically defined basal cells.
Mechanisms of Castration Resistance
Progressive prostate cancer, including primary disease and disease that occurs outside the prostate, is usually treated with ADT. [21] ADT almost always leads to reduced tumor burden, but ultimately the disease recurs in most cases. Accumulating evidence suggests that castration resistance can arise from one of many possible mechanisms, that maintain AR signaling. [22],[23],[24] AR-dependent mechanisms encompass approximately one-third of castration resistant prostate cancer (CRPC) and include gene amplifications or somatic mutations to AR and other pathway constituents. Such genetic changes usually occur in response to ADT, although rare pre-existing mutations have also been observed. [25],[26] An intrinsic mechanism of reduced androgen dependence following PTEN loss results from a self-reinforcing loop of depressed AR signaling and enhanced survival signaling. [27],[28] PTEN loss is associated with castration-resistant growth in mouse models and an increased probability of CRPC in patients. [29] Significant improvements have recently been incorporated into treatments that inhibit AR-dependent signaling, including the high-affinity AR antagonists MDV3100 and inhibitors of steroidal synthesis, such as abiraterone. [30] Nevertheless, it appears that over 50% of the patients do not respond or have partial responses to such enhanced ADT.
Lethal prostate cancers are heterogeneous and can include a range of AR expression, including cells that lack detectable expression. The initial hormone sensitivity of prostate cancers implies that androgen-independent cells are contained within a minor population. The scope of mechanisms that contribute to tumor re-growth is not clearly understood. Mechanisms that act independently of the androgen receptor might also play important roles. [31] Because castration-resistant regenerative stem/progenitor cells exist within normal prostates, one hypothesis is that phenotypically similar cells are present in progressive prostate cancer. An androgen-independent subpopulation within a heterogeneous prostate tumor could reflect the retained phenotype of a normal cell counterpart or, alternatively, could represent an acquired phenotype secondary to genetic changes [Figure 1]. Thus, one approach to analyzing mechanisms of castration resistance includes isolating and characterizing the hormone dependence and lineage phenotype of CSCs before and after ADT.
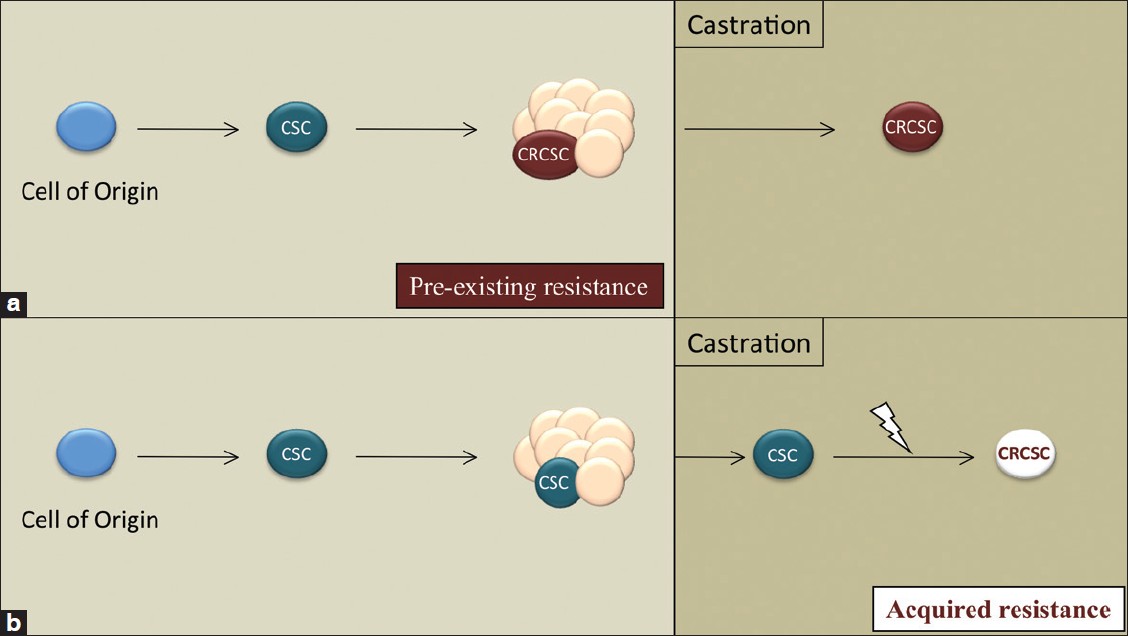 |
Figure 1: Hypothetical models for the evolution of a castration-resistant cancer stem cell (CSC). (a) Androgen independence may be a pre-existing feature of a subset of tumor cells including a CSC population. (b) Alternatively, CSCs may acquire castration resistance following additional genetic/epigenetic events. (a) and (b) are not mutually exclusive Click here to view |
Tumor Propagating Cells
The enrichment and molecular characterization of prostate cancer CSCs within heterogeneous tumor populations have significant translational potential. The identification of CSC-specific markers would allow histopathological analyses and correlation with clinical outcomes and might, ultimately, have prognostic utility. It has been proposed that combining standard debulking tumor therapy with CSC-directed therapy may improve the probability of avoiding therapy-resistant cancer. [32] The characterization of CSC signaling pathways, especially those mediating a survival function, could serve as effective therapeutic targets. Likewise, it is possible that CSCs represent a lineage-specific immature or “dedifferentiated” cell population and, therefore, targeted differentiation may provide a novel therapeutic approach. Finally, characterizing CSC populations before and after ADT would provide insight into the mechanisms of pre-existing and/or acquired androgen independence [Figure 1].
The currently accepted assay for delineating CSCs is based upon the ability of fractionated suspensions of tumor cells to form tumors of the appropriate histology following transplantation into immunocompromised mice. [3] As mentioned earlier, a major obstacle is the current inability to routinely grow freshly dissociated human prostate cells in the mouse. By contrast, the fractionation and transplantation of established human prostate cancer cell lines such as DU145 and PC3 have revealed the presence of subpopulations with enhanced tumor promoting activity. [33],[34] While useful, caution should be exercised when attempting to correlate data derived from such systems with candidate CSCs directly isolated from primary human tissue, as it is highly likely that substantial changes have occurred during adaption to and maintenance in tissue culture.
It is unclear as to why it has not been possible to perform CSC assays using primary prostate tumors. The frequently indolent or highly differentiated nature of bulk prostate cancer cells may contribute to weak transplantation ability. However, it is likely that various technical challenges contribute as well. A number of factors should be considered in this regard: the stage of tumor progression, the general health and viability of the cells used for transplantation, the length of time between surgical excision of the tumor and transplantation, and the protocol used to prepare the cells for transplantation. More difficult to determine, but still feasible, is the possibility that an important component of the human microenvironment is absent upon transplantation into the mouse. Nonetheless, efforts to address this significant problem are ongoing and should be encouraged, particularly given the recent compelling evidence demonstrating that alterations in tumor initiation assay conditions may have a major influence in the outcome of such studies. [4] Overcoming this issue is of critical importance to addressing the functional heterogeneity of human prostate cancer cell populations.
An alternative approach is to use model systems to investigate key questions concerning prostate cancer CSCs. Importantly, model systems begin to address the question of CSC frequencies. CSC markers discovered using model systems can be further investigated and validated with clinical samples. Newly identified cell surface markers have the potential to define additional subpopulations in both tumors and normal prostates. One model system that has recently been reported upon is serially transplanted human prostate xenograft tumors, CWR22 OT. CSCs defined by TRA-1-60, CD151 and CD166 expression were characterized as immature AR-negative cells with an intermediate cytokeratin profile and lacking in the definitive basal marker, TP63. [35] These data support the presence of multipotent CSCs in human prostate cancers and suggest that it will be worthwhile to investigate additional primary prostate cancer xenografts.
A complimentary system takes advantage of genetically engineered mouse models (GEMMs) of prostate cancer. The availability of fresh tumor tissue and the ability to readily manipulate the hormone environment of tumor-bearing mice are of obvious benefit. Data using mouse models are still limited. Using the PbCre4; Pten fl/fl model, Mulholland et al. demonstrated that a primary tumor fraction enriched for basal cells produced upon renal graft transplantation neoplastic glands containing basal and luminal cells. [36] It is unclear how closely related the CSC described here is to an adenocarcinoma CSC. The identification and evaluation of models giving rise to transplantable adenocarcinoma are of obvious interest.
Key factors in the optimal use of GEMMs are evaluating the pathologically comparable features of the mouse and human tumors and interpreting the modeling strategy. The combination of genetic changes comprising the oncogenic stimulus and the cellular populations in which the genetic changes occur (i.e., the cells of origin) are significant factors in relating mechanisms of transformation between the two species [Figure 2]. As has been shown in the hematopoietic [37],[38],[39] and mammary [40] systems previously, the oncogenic event itself may dictate the cell type transformed. More recently, in a study of lung cancer GEMMs, Curtis et al. determined that the cell surface phenotype of the lung CSCs responsible for adenocarcinogenesis was dependent on the combination of genetic changes driving the malignancy. [41] Below, we briefly discuss the current understanding of prostate cancer cells of origin, and we highlight the considerations regarding modeling genetic changes.
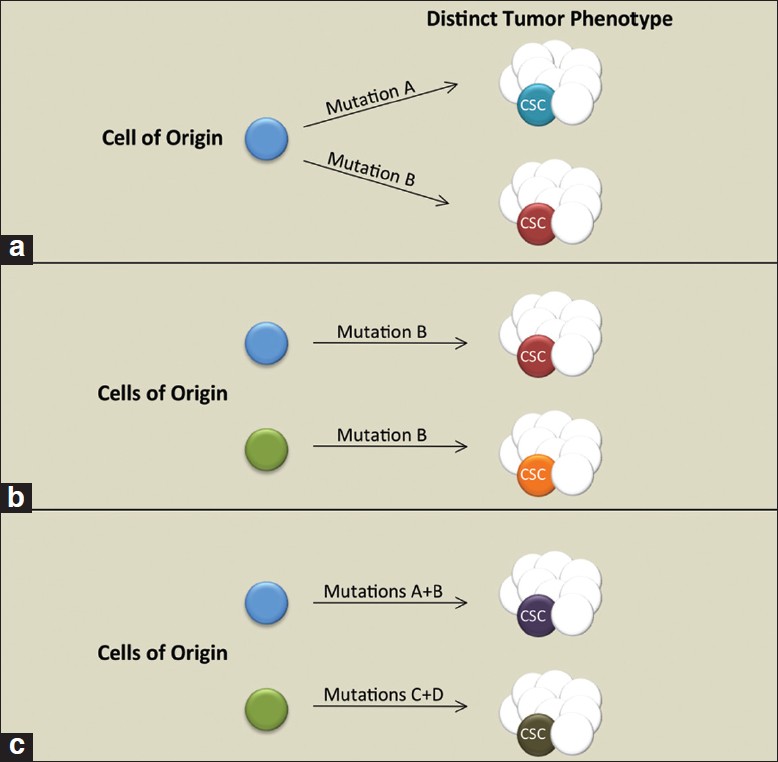 |
Figure 2: Contribution of cell type and oncogenic events in dictating tumor and cancer stem cell (CSC) phenotype. (a) Separate oncogenic events dictate tumor and CSC phenotype. (b) Transformation of distinct cell lineages influences tumor/ CSC phenotype. (c) A combination of oncogenic events and the targeted cell lineage impacts tumor and CSC phenotype Click here to view |
Cell of Origin
The defining property of a candidate cell of origin is its susceptibility to oncogenic transformation. The cell of origin is considered to be a normal cell within a tissue that receives the first cancer promoting mutation(s). [42] As such, it may differ significantly from the cell or subset of cells responsible for sustaining tumor growth, namely the CSC [Figure 2]. Two major approaches have been used to identify cells of origin: cell type-specific promoters driving an oncogenic stimulus or FACS isolation of cell populations followed by ex vivo genetic alterations and transplantation of the cells.
Mouse models
The data collected from mouse modeling studies are consistent with the initiating mutations for prostate adenocarcinoma occurring in either the luminal or the multipotent stem/progenitor compartments. Comparisons between models have been possible because the effects of a common mutation, deletion of floxed Pten alleles, were initiated by promoter-specific cre-recombinase (CRE) expression. Transgenic CRE expression driven by a composite probasin promoter, PB-Cre4, resulted in PIN lesions containing proliferating basal cells and their differentiating intermediate cell progeny. [43] Adenocarcinoma was observed temporally following PIN development, suggesting progression. These data imply that lesions developed from multipotent progenitors that ultimately differentiated to a luminal phenotype. This interpretation is supported by PB-Cre4-dependent deletion of Pten and TP53. In this model, clonal multipotent progenitor cell lines that give rise to adenocarcinoma were isolated from prostate tumors. [44] Although probasin is generally expressed in luminal cells, it appears that in the Pb-Cre4 transgenic line, sufficient CRE expression occurs to establish Pten deletion in self-renewing cells with the plasticity to produce both luminal and TP63+ basal cells. In a different model, Ma and colleagues used PSA promoter-driven Cre recombinase deletion of Pten, which was limited to luminal cells. [45],[46] There was development of PIN and adenocarcinoma, which differed from PB-Cre4-initiated disease in having a slower time course and no obvious involvement of transformed basal cells. Therefore, luminal lineage cells can be targets for adenocarcinoma development. In another luminal cell model, castration-resistant luminal cells with regenerative potential, referred to as CARNs, have been shown to initiate PIN/adenocarcinoma following Pten deletion. [13] Of the various mouse models, CARNs and their progeny are the only cells marked using genetic lineage tracing and, thus, formally shown to be targets of transformation. Outstanding questions with respect to CARNs are whether they exist within non-castrated prostates and, if so, whether they can serve as a cell of origin in such an environment?
Cell fractionation
A second strategy for investigating transformation targets in prostate epithelial cells utilizes cell surface markers and flow cytometry to separate cell populations, followed by ex vivo infection with lentiviruses encoding oncogenic stimuli. Minimally cultured, infected cells are subsequently transplanted in the presence of mesenchyme to evaluate the neoplastic characteristics of the outgrowths that develop. Overall, this approach identified cells within the basal/stem cell fraction as giving rise to PIN/adenocarcinoma. Studies from Owen Witte’s laboratory have shown that transformation of basal/stem-enriched cell fractions with constitutively active AKT in mouse cells [47] or AKT and ERG in human cells [48] led to PIN-like growths. Because developing glandular structures have been shown to be clonal in origin, [10] these data imply that PIN develops from an initiating cell with the capacity to produce basal and luminal cells. Interestingly, the addition of ectopic AR expression in the above transformants led to adenocarcinomas with loss of basal cells. This suggests the interesting possibility that AR signaling stimulates differentiation to a transformation-susceptible cell that has lost basal differentiation capacity. Alternatively, increased AR signaling may be an early step in the transformation process, and ectopic expression enhances the efficiency of progression, i.e. adenocarcinoma development. Finally, although the fractionation approach has demonstrated that transformation of a basal/stem cell can lead to adenocarcinoma, the absence of activity within the luminal fraction is harder to interpret because a negative result may be due to technical problems.
Cells of Origin and Cancer Stem Cells
Taken together, the approaches described above suggest that transformation of luminal or multipotent progenitors are capable of giving rise to adenocarcinomas. Transformation of luminal cells gives rise to disease that more closely resembles the pathology of clinically relevant, human prostate cancer. On the other hand, the rapid, polyclonal nature of prostate cancer development in models such as PB-Cre4-induced Pten deletion may amplify occurrences, such as multipotent progenitor proliferation, which represent early, rare, subclinical transformation events in humans. A significant question is whether adenocarcinomas initiated from distinct cells of origin converge on a common CSC [Figure 2]. Future studies aimed at systematic comparisons of the prostate CSCs in mouse models will determine how genetic alterations and cell of origin targeting influence their phenotype, including castration resistance. Oncogenotype regulation of CSC populations may have implications for tailoring treatment in human prostate cancer patients.
The Future of Cell Surface Markers in Prostate CSC Research
The use of cell surface markers coupled with FACS has proved a central tool for the isolation of multiple candidate CSC populations. Interestingly, many CSC populations express similar markers (for example CD133) even when isolated from different tissues. However, increasing evidence suggests that surface markers presently used may not be exclusive to CSC populations. [49] This may require a reassessment of our expectations with respect to the use of such markers. To date, few markers have been identified that are exclusive to CSC populations. As cell surface markers will undoubtedly remain a mainstay of CSC research, it may be more practical to look at CSC surface marker expression in terms of level of expression and in combination with multiple other markers, rather than simply presence or absence. The significant advances in multicolor flow cytometry now allows for multiple markers to be assessed simultaneously, which in turn will allow a cumulative profile of multiple markers to be generated for each CSC population. The combined use of markers has been effective in isolating hematopoietic cell lineages. [50] There is a paucity of characterized surface markers to isolate prostate epithelial populations, which has resulted in enriched but significantly heterogeneous cell fractions. Future efforts to identify and characterize additional surface markers and experimental approaches to purify populations are crucial for optimal progress.
With respect to prostate cancer, the ongoing debate regarding the phenotype of cells responsible for cancer initiation and tumor propagation is predicated on a handful of intracellular differentiation markers, including CK5, p63, CK8 and Nkx3.1. It is possible that the current markers do not reveal every individual prostate cell lineage, especially following oncogenic events. Also, multiple intermediate/progenitor cells may go undetected if their marker expression is transient. As our ability to more finely tag and parse the prostate epithelial cell compartment improves, it is likely that more subpopulations of cells relative to prostate cancer will appear. We need only look at the progression of the lineage map of the hematopoietic system to be reminded of the complexity of differentiating biological systems.
Conclusions and Future Directions
The development of concepts and techniques to address tumor heterogeneity in various cancers has refined our thinking about the need to identify prognostic subpopulations of tumor cells and about treatment paradigms to co-target cells of different differentiation status in tumors. Although transplantation conditions for primary human prostate cancers have not been developed to date, model systems are available to begin analyzing fundamental questions concerning the phenotype of prostate CSCs. Do adenocarcinomas contain a definable subpopulation of CSCs? If so, what are their unique molecular markers? Are such cells capable of surviving castration and propagating castration-resistant disease. Are murine CSCs across all models created equal? Does the oncogenotype affect the cell responsible for tumor initiation? The answers to these and other related questions are certain to influence basic and translation research into the diagnosis and treatment of prostate cancer.
Acknowledgments
The authors would like to thank members of the Kelly lab for useful advice and discussion.
References
| 1. | American Cancer Society: Cancer Facts and Figures 2011. In.; 2011. http://www.cancer.org  |
| 2. | Alison MR, Lim SM, Nicholson LJ. Cancer stem cells: problems for therapy? J Pathol 2011;223:147-61. [PUBMED] [FULLTEXT] |
| 3. | Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med 2011;17:313-9. [PUBMED] [FULLTEXT] |
| 4. | Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature 2008;456:593-8. [PUBMED] [FULLTEXT] |
| 5. | Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 2010;140:62-73. [PUBMED] [FULLTEXT] |
| 6. | Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009;138:592-603. [PUBMED] [FULLTEXT] |
| 7. | Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev 2000;14:2410-34. [PUBMED] [FULLTEXT] |
| 8. | Isaacs JT. Control of cell proliferation and cell death in the normal and neoplastic prostate: a stem cell model. In: Benign Prostatic Hyperplasia. In: Rodgers CH, Coffey DS, Cunha G, Grayshack R, Henman J, Horton R, editors. Vol. 87. 1985. p. 2881. |
| 9. | Tsujimura A, Koikawa Y, Salm S, Takao T, Coetzee S, Moscatelli D, et al. Proximal location of mouse prostate epithelial stem cells: a model of prostatic homeostasis. J Cell Biol 2002;157:1257-65. [PUBMED] [FULLTEXT] |
| 10. | Xin L, Lawson DA, Witte ON. The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc Natl Acad Sci U S A 2005;102:6942-7. [PUBMED] [FULLTEXT] |
| 11. | Burger PE, Xiong X, Coetzee S, Salm SN, Moscatelli D, Goto K, et al. Sca-1 expression identifies stem cells in the proximal region of prostatic ducts with high capacity to reconstitute prostatic tissue. Proc Natl Acad Sci U S A 2005;102:7180-5. [PUBMED] [FULLTEXT] |
| 12. | Leong KG, Wang BE, Johnson L, Gao WQ. Generation of a prostate from a single adult stem cell. Nature 2008;456:804-8. [PUBMED] [FULLTEXT] |
| 13. | Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009;461:495-500. |
| 14. | Bostwick DG. Prostatic intraepithelial neoplasia. Urology 1989;34(6 Suppl):16-22. |
| 15. | DeMarzo AM, Nelson WG, Isaacs WB, Epstein JI. Pathological and molecular aspects of prostate cancer. Lancet 2003;361:955-64. [PUBMED] [FULLTEXT] |
| 16. | Nadig SN, Deibler AR, El Salamony TM, Hull GW, Bissada NK. Small cell carcinoma of the prostate: an underrecognized entity. Can J Urol 2001;8:1207-10. [PUBMED] |
| 17. | Grignon DJ. Unusual subtypes of prostate cancer. Mod Pathol 2004;17:316-27. [PUBMED] [FULLTEXT] |
| 18. | Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol 2008;21:1156-67. [PUBMED] [FULLTEXT] |
| 19. | Meeker AK, Hicks JL, Platz EA, March GE, Bennett CJ, Delannoy MJ, et al. Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res 2002;62:6405-9. [PUBMED] [FULLTEXT] |
| 20. | De Marzo AM, Nelson WG, Bieberich CJ, Yegnasubramanian S. Prostate cancer: New answers prompt new questions regarding cell of origin. Nat Rev Urol 2010;7:650-2. [PUBMED] [FULLTEXT] |
| 21. | Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 2008;8:440-8. [PUBMED] [FULLTEXT] |
| 22. | Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004;10:33-9. [PUBMED] [FULLTEXT] |
| 23. | Mellado B, Codony J, Ribal MJ, Visa L, Gascon P. Molecular biology of androgen-independent prostate cancer: the role of the androgen receptor pathway. Clin Transl Oncol 2009;11:5-10. |
| 24. | Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol 2009;6:76-85. [PUBMED] [FULLTEXT] |
| 25. | Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18:11-22. [PUBMED] [FULLTEXT] |
| 26. | Bergerat JP, Ceraline J. Pleiotropic functional properties of androgen receptor mutants in prostate cancer. Hum Mutat 2009;30:145-57. |
| 27. | Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011;19:575-86. [PUBMED] [FULLTEXT] |
| 28. | Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011;19:792-804. [PUBMED] [FULLTEXT] |
| 29. | Sircar K, Yoshimoto M, Monzon FA, Koumakpayi IH, Katz RL, Khanna A, et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J Pathol 2009;218:505-13. [PUBMED] [FULLTEXT] |
| 30. | Yap TA, Zivi A, Omlin A, de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol 2011;8:597-610. |
| 31. | Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med 2010;16:1414-20. [PUBMED] [FULLTEXT] |
| 32. | Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 2011;108:7950-5. [PUBMED] [FULLTEXT] |
| 33. | Wei C, Guomin W, Yujun L, Ruizhe Q. Cancer stem-like cells in human prostate carcinoma cells DU145: the seeds of the cell line? Cancer Biol Ther 2007;6:763-8. [PUBMED] [FULLTEXT] |
| 34. | Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, García-Echeverría C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A 2009;106:268-73. |
| 35. | Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat Commun 2011;2:162. [PUBMED] [FULLTEXT] |
| 36. | Mulholland DJ, Xin L, Morim A, Lawson D, Witte O, Wu H. Lin-Sca-1+CD49fhigh stem/progenitors are tumor-initiating cells in the Pten-null prostate cancer model. Cancer Res 2009;69:8555-62. |
| 37. | Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 2004;119:431-43. [PUBMED] [FULLTEXT] |
| 38. | Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004;6:587-96. [PUBMED] [FULLTEXT] |
| 39. | Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006;442:818-22. [PUBMED] [FULLTEXT] |
| 40. | Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 2009;15:907-13. [PUBMED] [FULLTEXT] |
| 41. | Curtis SJ, Sinkevicius KW, Li D, Lau AN, Roach RR, Zamponi R, et al. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell 2010;7:127-33. [PUBMED] [FULLTEXT] |
| 42. | Visvader JE. Cells of origin in cancer. Nature 2011;469:314-22. [PUBMED] [FULLTEXT] |
| 43. | Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U S A 2006;103:1480-5. [PUBMED] [FULLTEXT] |
| 44. | Martin P, Liu YN, Pierce R, Abou-Kheir W, Casey O, Seng V, et al. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am J Pathol 2011;179:422-35. [PUBMED] [FULLTEXT] |
| 45. | Ma X, Ziel-van der Made AC, Autar B, van der Korput HA, Vermeij M, van Duijn P, et al. Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res 2005;65:5730-9. [PUBMED] [FULLTEXT] |
| 46. | Korsten H, Ziel-van der Made A, Ma X, van der Kwast T, Trapman J. Accumulating progenitor cells in the luminal epithelial cell layer are candidate tumor initiating cells in a Pten knockout mouse prostate cancer model. PLoS One 2009;4:e5662. [PUBMED] [FULLTEXT] |
| 47. | Lawson DA, Zong Y, Memarzadeh S, Xin L, Huang J, Witte ON. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc Natl Acad Sci U S A 2010;107:2610-5. [PUBMED] [FULLTEXT] |
| 48. | Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science 2010;329:568-71. [PUBMED] [FULLTEXT] |
| 49. | Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J Clin Invest 2008;118:2111-20. [PUBMED] [FULLTEXT] |
| 50. | Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005;121:1109-21. [PUBMED] [FULLTEXT] |
Authors
Dr. Paul G. Hynes, Cell and Cancer Biology Branch, National Cancer Institute, 37 Convent Drive, Rm 1066, Bethesda, MD
Dr. Kathleen Kelly, National Institutes of Health 37 Convent Drive, Room 1068 Bethesda, MD
Figures