Natalia A Ignatenko1, Eugene W Gerner1, David G Besselsen2
1 Department of Cell Biology and Anatomy, Arizona Cancer Center, 1515 N. Campbell Ave., Tucson, Arizona 85724, USA
2 University Animal Care, University of Arizona Central Animal Facility, Room 116, 1127 E Lowell St, Tucson, AZ 85721-0101, USA
| Date of Submission | 24-Jan-2011 |
| Date of Acceptance | 05-Feb-2011 |
| Date of Web Publication | 16-Apr-2011 |
Correspondence Address:
Natalia A Ignatenko
Department of Cell Biology and Anatomy, Arizona Cancer Center, 1515 N. Campbell Ave., Tucson, Arizona 85724
USA
Source of Support: None, Conflict of Interest: None
DOI: 10.4103/1477-3163.79673
Abstract
Genetics and diet are both considered important risk determinants for colorectal cancer, a leading cause of death in the US and worldwide. Genetically engineered mouse (GEM) models have made a significant contribution to the characterization of colorectal cancer risk factors. Reliable, reproducible, and clinically relevant animal models help in the identification of the molecular events associated with disease progression and in the development of effictive treatment strategies. This review is focused on the use of mouse models for studying the role of polyamines in colon carcinogenesis. We describe how the available mouse models of colon cancer such as the multiple intestinal neoplasia (Min) mice and knockout genetic models facilitate understanding of the role of polyamines in colon carcinogenesis and help in the development of a rational strategy for colon cancer chemoprevention.
Keywords: Apc Min/+ , DFMO, mouse models, NSAIDs, polyamines, SLC3A2
How to cite this article:
Ignatenko NA, Gerner EW, Besselsen DG. Defining the role of polyamines in colon carcinogenesis using mouse models. J Carcinog 2011;10:10
How to cite this URL:
Ignatenko NA, Gerner EW, Besselsen DG. Defining the role of polyamines in colon carcinogenesis using mouse models. J Carcinog [serial online] 2011 [cited 2021 Oct 15];10:10. Available from: https://carcinogenesis.com/text.asp?2011/10/1/10/79673
Introduction
The polyamines (diamine putrescine, triamine spermidine, and tetraamine spermine) are biogenic amines that are widely distributed in living organisms. The compounds have low molecular weight and are positively charged at physiological pH values, which allow them to interact with any negatively charged cellular component. These polycations have diverse biological functions, including transcription, RNA stabilization and translational frameshifting, ion channel gating, and other processes. [1],[2],[3] Polyamine levels within cells control cell viability, and significant change in polyamine level results in apoptosis. [4] Polyamine homeostasis in cells is achieved by the regulatory mechanisms of synthesis, degradation, and membrane polyamine transport. [5] The intracellular polyamine homeostasis is lost during the process of cancer development as shown by the upregulation of polyamine biosynthetic enzymes, with corresponding decrease in polyamine catabolism, in these conditions. [6],[7] Studies over many years using human colon cancer cell lines and genetically engineered mouse (GEM) models have generated evidence that ornithine decarboxylase (ODC), the first enzyme in polyamine synthesis, is upregulated during carcinogenesis. [6] Suppression of polyamine biosynthesis with an inhibitor α-difluoromethylornithine (DFMO) suppressed cancer development in animal models and in human clinical trials.[8],[9] This brief review highlights in vivo studies that have focused on the role of polyamines in colon carcinogenesis and the manipulations of the polyamine pathway by dietary and pharmaceutical methods to maintain healthy homeostasis.
Gems for the Study of the Regulation of Polyamine Metabolism
Polyamine biosynthesis is controlled by ornithine decarboxylase (ODC), which catalyses the formation of putrescine from the amine acid ornithine. Putrescine is subsequently converted into spermidine through the actions of S-adenosylmethionine decarboxylase (AMD1) and spermidine synthase (SPDS). Spermidine is then converted into spermine by AMD1 and spermine synthase (SMS). Important catabolic effectors maintain polyamine homeostasis as well. Spermidine/spermine N-acetyltransferase (SAT1) adds terminal acetyl groups to spermidine and spermine, which accelerates the export of acetylated polyamines. Spermine oxidase (SMO) converts spermine to spermidine. Acetylpolyamine oxidase (PAOX) also aids in polyamine homeostasis by converting acetylated spermidine and spermine back to putrescine and spermidine, respectively [Figure 1].
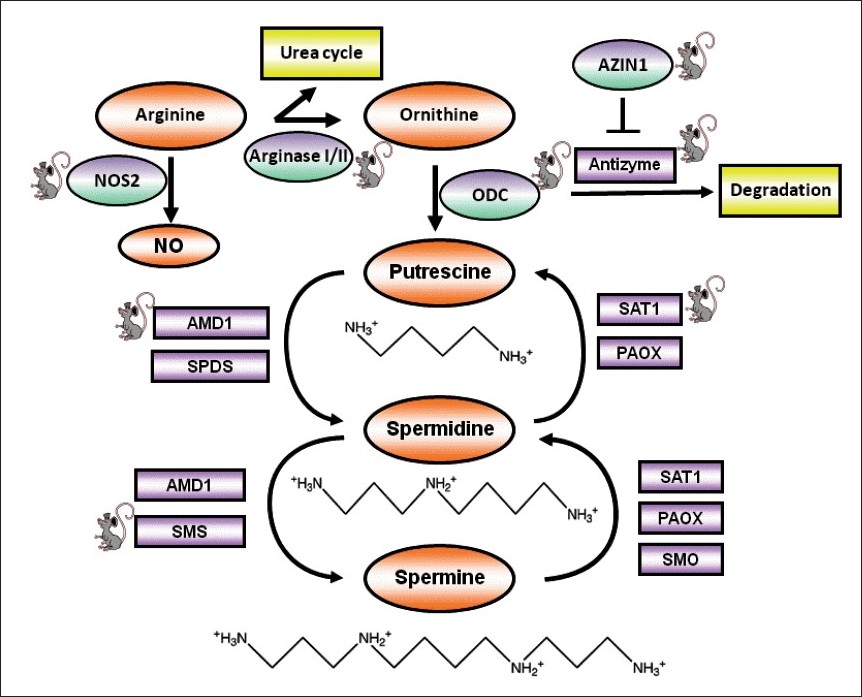 |
Figure 1: Polyamine structure and metabolism. Enzymes are shown in purple squares, except for ODC, NOS2, AZIN1, and arginase I/II, which are shown in purple ovals. (ODC, ornithine decarboxylase; NOS2, nitric synthase 2; AZIN1, antizyme inhibitor 1; AMD1, S-adenosylmethionine decarboxylase; SAT1, spermidine/spermine N1-acetyltransferase; SMO, spermine oxidase; PAOX, acetylpolyamine oxidase). Mouse icons represent GEM models that have been mentioned in the text. Click here to view |
Mouse models of polyamine synthesis
Multiple studies have demonstrated that ODC is an essential metabolic effector required for normal development in mammals. ODC gene knockouts are lethal in murine embryos 3.5 days after fertilization. [10] Overexpression of ODC in a transgenic mouse resulted in accumulation of putrescine but had much less effect on higher polyamines, indicating that polyamine metabolism remains under homeostatic control. [11] ODC transcription is activated in response to hormones, growth factors, and tumor promoters via the response elements in the ODC gene promoter. [12] Of special interest, the ODC gene promoter contains three enhancer (E) boxes (CACGTG) that are binding sites for the MYC, MAX, MAD, and MNT transcription factors. [13]
ODC protein levels in cells are regulated by an ODC inhibitory protein called ornithine decarboxylase antizyme (OAZ). [14],[15] Full-length functional antizyme acts as a noncompetitive inhibitor of ODC and enhances ubiquitin-independent ODC degradation by the 26S proteasome. [14] In addition, antizyme increases polyamine efflux and suppresses polyamine uptake, thus decreasing the intracellular polyamine pool. [16],[17] The antizyme transgenic mouse models with increased antizyme expression from the bovine keratin 5 (K5) or keratin 6 (K6) promoters develop normally and are phenotypically indistinguishable from wild-type littermates. [18]
Antizyme is regulated by the antizyme inhibitor 1 (AZIN1), which is a homolog of ODC but lacks the decarboxylation activity. [19] AZIN1 sequesters the intracellular pool of antizyme, which causes an increase in the levels of ODC protein, ODC activity, and polyamine levels. An AzIn1 knockout mouse was developed and analyzed using Agilent oligonucleotide array. [20] Genes found to be differentially expressed in the AzIn1 knockout model were involved in polyamine metabolism, embryonic morphogenesis, regulation of cell cycle and proliferation, signal transduction cascades, immune response, and apoptosis.
AMD1 converts putrescine into the higher polyamines by producing the aminopropyl donor decarboxylated S-adenozylmethionine (dcSAM). The activity of AMD1 is highly regulated at the level of transcription, translation, and protein turnover. [21] The AMD1 gene contains a number of binding sites for different transcription factors, including a spermidine response element. Putrescine activates mammalian AMD1 by enhancing the production of the processed form of the enzyme and improving its catalytic activity. [21] Protein ubiquitination has also been shown to regulate AMD1 turnover. [22],[23] Targeted disruption of the mouse Amd1 gene showed that, similarly to ODC, homozygous knockout of Amd1 is lethal in murine embryos past E3.5, which indicates that AMD1 plays an essential role in embryonic development and that spermidine and spermine are required for embryonic development after E3.5. [24] Mice overexpressing Amd1 did not show any significant changes in the tissue levels of spermidine and spermine due to their acetylation and export. [25]
Mammalian spermidine and spermine synthase are regulated by the availability of putrescine or spermidine and dcSAM. Recent studies by Forshell et al.[26] have described the induction of spermidine synthase gene transcription by the c-MYC oncogene in a murine model of B-cell lymphoma. The putative c-MYC binding site is associated with canonic E-boxes located upstream and downstream of exon 1 in the SMS gene. The consequences of the deletion of SMS have been tested in vivo using Gyro (GY) mice, which have a deletion of the X chromosome that eliminates the SMS gene.[27] The SMS knockout mice have multiple abnormalities, including a tendency to sudden death, small size, hearing loss, lack of fertility, circling behavior, and other neurological symptoms. The hearing loss in Gy mice was reversed by breeding them with CAG/SmS mice, a transgenic line that ubiquitously expresses spermine synthase under the control of a composite cytomegalovirus-IE enhancer/chicken β-actin promoter. [28],[29] Treatment of Gy mice with the ODC inhibitor DFMO resulted in their death within 5 days of exposure to the drug due to loss of motor function.[29] This finding emphasizes the critical role of polyamines in the regulation of Kir channels that maintain the endocochlear potential in the inner ear and the importance of a normal spermidine/spermine ratio in hearing and balance functions.
Mouse models of polyamine catabolism
A key polyamine catabolic enzyme SAT1 is highly inducible by a variety of stimuli, including elevated polyamine levels, synthetic polyamine analogs, toxins, hormones, cytokines, heat shock, ischemia-reperfusion injuries, and other stresses. [30] The regulation of SAT1 occurs via activation of transcription, mRNA processing, mRNA translation, and protein turnover. [30],[31],[32],[33] The SAT1 gene contains the binding sites for common regulators of gene expression such as SP1 and AP1, CCAAT/enhancer-binding protein βC/EBPβ), cAMP-response-element-binding protein (CREB), nuclear factor κB (NF-κB), and peroxisome-proliferator-activated receptors (PPARs) transcription factors. SAT1 also contains a polyamine-response element (PRE), which binds polyamines and polyamine-responsive transcription factors. [34] The posttranscriptional regulation of SAT1 involves regulation of mRNA stability, alternative splicing of SAT1 mRNA, translational regulation of SAT1 synthesis, and stabilization of SAT1 protein; this has been reviewed elsewhere. [35] The Sat1 knockout mice were generated via a targeted inactivating insertion within the X-linked Sat1 gene. [36],[37] Sat1 knockout mice are viable, fertile, and show no obvious change in phenotype. The overexpression of Sat1 in transgenic mice has been achieved under the control of an endogenous promoter as well as the heavy metal-inducible metallothionin promoter.[38],[39] Significant phenotypic changes were found in these mice, such as substantial alterations in the polyamine pool, early and permanent hair loss, female infertility, weight loss, and lack of subcutaneous fat. [39] Sat1 overexpression also led to the disappearance of visceral fat depots and general lipoatrophy.[40],[41]
Polyamine metabolism also depends on the levels of the precursor amino acids arginine and ornithine. Both amines, as well as putrescine and other polyamines, are available in the normal diet. [42],[43] The role of arginase enzymes (arginase I and arginase II isoforms), which produce ornithine that is utilized for the polyamine synthesis, and therefore could be important for the maintenance of the polyamine pathway, has been assessed in vivo using arginase knockout mice. [44] The disruption of arginase I (AI−/− ) and arginase II (AII−/− ) did not greatly affect the ODC enzyme activity and polyamine levels, suggesting that endogenous arginase does not directly contribute to polyamine homeostasis in mice. Thus, exogenous sources of arginine play the more significant role in modulating polyamine levels, as will be discussed further.
Colon Cancer Risk
Colon cancer is the fourth most commonly diagnosed cancer and the second leading cause of cancer-related death in the US. [45] A key contributor to colon cancer risk is heredity. Molecular genetic studies have identified key genes whose mutations or altered expression can lead to colon cancer. The mutated genes are found in two key pathways: the APC/Wnt pathway and the TGFβ pathway. The APC gene, a tumor-suppressor gene located on chromosome 5, is found mutated in the early stages of colon carcinogenesis and is linked to familial adenomatous polyposis (FAP). Mutations in the DNA mismatch repair genes (MSH2, MLH1, PMS1, or PMS2 genes) lead to hereditary nonpolyposis colon cancer (HNPCC) and frequently result in mutations of genes within the TGF-β pathway. [46],[47] In addition, mutations in the K-RAS oncogene are found in 50% of intermediate and late adenomas and facilitate the progression of adenomas to neoplasia. [48] The p53 tumor-suppressor gene is associated with the invasive phenotype of colon tumors, as evidenced by a high rate of mutations within late adenomas and colorectal cancers. [49] Substantial evidence exists that diet and physical activity also are important determinants of colon cancer risk. Dietary components, such as red meat (including beef, pork, lamb, and goat), processed meat, and alcohol have been associated with increased colon cancer risk. [50],[51],[52]
Effect of genetic risk factors of colon carcinogenesis on the polyamine pathway in vivo
Mutations in the APC gene are necessary to initiate FAP and are also important in sporadic colorectal tumorigenesis. The multiple intestinal neoplasia (Min or Apc Min/+) mouse contains a truncating mutation at codon 850 of the Apc gene and develops an average of 30-60 macroscopically visible pan-intestinal and desmoid tumors and, infrequently, colonic tumors. [53],[54] In contrast, human FAP patients develop hundreds of, predominantly colonic, adenomas that progress to invasive adenocarcinomas by 40 years of age. [55] We have reported the altered polyamine metabolism in the Apc Min/+ mice. [56] The increased polyamine levels in the small intestine of the Apc Min/+ mice were associated with an increase in ODC and decrease in AZ steady-state RNA levels. We also found that the treatment of Apc Min/+ mice with a specific ODC inhibitor, DFMO, suppressed the intestinal polyamine levels and the APC-dependent intestinal tumorigenesis in the Apc Min/+ mice, thus providing direct evidence that polyamines are the mediators of the mutant APC-initiated tumorigenesis. Since the APC tumor-suppressor gene is mutated at the early stages of colon carcinogenesis, the Apc Min/+ mouse has been utilized for evaluation of the contribution of polyamine metabolic genes in colon carcinogenesis [Table 1]. In vitro experimental studies using human colon tumor cell lines showed that wild-type APC suppresses the expression of the c-Myc oncogene, which activates the ODC gene and, therefore, polyamine biosynthesis. [57] We have reported that suppression of c-Myc expression in vivo, using the mouse model with a conditional deletion of c-MYC in the intestinal and colonic mucosa, resulted in a decrease of mutant APC-dependent intestinal tumorigenesis. [58] Our data suggests that c-MYC plays an important role in the development of intestinal tumors with activating mutations in the APC gene, and that this role is associated with decreased apoptosis in the normal intestinal epithelium.
| Table 1: Mouse models used for analysis of the role of polyamine pathway in colon carcinogenesis Click here to view |
The role of SAT1 in intestinal tumorigenesis has been tested by crossing the Apc Min/+ mice with either transgenic Sat1 mice or Sat1 knockout mice. [37] The finding of this study was that the number of adenomas in both the small intestine and colon were increased in Apc Min/+ ; Sat1 Tg relative to Apc Min/+ ; Sat1 +/+ mice. This was associated with an increase in ODC and AMD enzyme activities and putrescine and N1-acetylspermidine levels, but the levels of spermidine and spermine were not changed. [37] Accordingly, crossing the Apc Min/+ mice with an Sat1-deficient mouse strain resulted in 75% reduction in adenoma numbers in the small intestine. [37] This analysis indicates that SAT1 promotes tumor development in Apc Min/+ mice. The observed enhanced intestinal and colonic tumorigenesis in the Sat1-overexpressing Apc Min/+ mice appears to be contradictory with the established role of SAT1 as a polyamine catabolic enzyme acting to reduce the intracellular polyamine concentrations. Sat1 overexpression resulted in inhibition of tumorigenesis in some mouse cancer models, such as the TRAMP mouse model of prostate cancer and the two-stage skin carcinogenesis model. [40],[59] The differential impact of SAT1 on tumorigenesis in different mouse models has been attributed to the tissue-specific alterations in polyamine pools caused by activated catabolism and compensatory increase in polyamine biosynthesis to maintain the polyamine homeostasis; [60] however, the detailed mechanism has not yet been defined.
Assessment of the role of dietary polyamines in colon carcinogenesis and in response to chemopreventive agents in vivo
Role of dietary arginine in colon carcinogenesis
Arginine is the substrate for nitric oxide synthases (NOSs), which breaks it down to produce nitric oxide (NO) and citrulline. Arginine is also catabolized by the enzyme arginase to form ornithine, which is necessary for polyamine synthesis and thus influences intracellular polyamine levels. We have evaluated the role of polyamines in intestinal and colon carcinogenesis in Apc Min/+ mice fed with dietary supplements of arginine. [61] The concentrations of dietary arginine supplementation used (0.2% and 2% in drinking water) were comparable to the dietary consumption of arginine in humans. We found that dietary arginine supplementation enhanced colon, but not intestinal, tumor incidence and adenoma grade in the Apc Min/+ mice [62] [Table 2]. Deletion of the inducible nitric oxide synthase gene (Nos2), which is the major contributor to the production of NO from arginine, resulted in decreased tumorigenesis in both small intestine and colon. Treatment of Apc Min/+ mice with DFMO suppressed the arginine-dependent increase in the colon tumor burden and grade. [61] These data provide experimental evidence that dietary arginine is a luminal risk factor for colon carcinogenesis. The major source of dietary arginine in humans is meat, with high arginine quantities being found in red meat, pork, and chicken. [42] Epidemiological studies on the effects of estimated arginine exposures through meat consumption on tumor characteristics and survival in colorectal cancer patients showed that patients with familial colorectal cancers in the highest meat consumption quartile had decreased overall survival, an effect that was shown to be independent on adjusted regression analysis. [63]
| Table 2: Role of dietary polyamines in colon carcinogenesis in ApcMin/+ mouse model Click here to view |
Effect of dietary polyamines in colon carcinogenesis in vivo
Besides arginine, dietary polyamines have also been found to enhance intestinal and colonic tumorigenesis. [64] Studies by our group indicate that Apc Min/+ mice given dietary supplements of putrescine (1% in the drinking water) show an increase in adenoma grade [65] [Table 2].
The molecular targets for the antitumor activity of the nonsteroidal anti-inflammatory drugs (NSAIDs) sulindac and celecoxib within the polyamine pathway have been evaluated using experimental animal models, including Apc Min/+ mice. [65] Specifically, sulindac increased steady-state RNA levels and enzymatic activity of the polyamine catabolic enzyme SAT1. Sulindac also decreased the activity of the biosynthetic enzyme ODC, but not that of AMD1. The effectiveness of sulindac in suppressing intestinal carcinogenesis was partially abrogated by dietary putrescine. [65] Thus, dietary polyamines can modulate the effectiveness of colon cancer preventive agents. Since high concentrations of putrescine can be found in certain dietary components, it may be advantageous to restrict dietary putrescine consumption in patients undergoing treatment with sulindac. The combination of DFMO with sulindac, a nonselective inhibitor of cyclooxygenases 1 and 2 (COX-1 and COX-2), or celecoxib, a selective COX-2 inhibitor, was additive in suppressing tumorigenesis in Apc Min/+ mice. [63] At the same time, the combination of DFMO with sulindac in the Apc Min/+ mouse model was more effective than the combination of DFMO with celecoxib in reducing both the intestinal polyamine content and the incidence of high-grade intestinal adenomas. [66] These positive experimental data provided a rational for further evaluation of the cancer chemopreventive efficacy of the combination of DFMO and sulindac in a phase IIb/III clinical trial for the prevention of colon polyp recurrence in patients with sporadic colorectal adenomas. [9] The DFMO and sulindac combination in this trial was associated with a dramatic 92% reduction in advanced colorectal adenomas.
Role of polyamine transport in colon carcinogenesis in vivo
Extracellular sources of polyamines include foods high in polyamines, such as cheese and meat, [43] and polyamines produced by intestinal bacteria. Previous work from our laboratory has shown that polyamine transport into cells occurs by a caveolae-dependent mechanism. [67] Since ornithine and arginine are involved in important metabolic pathways, the supply of these amino acids is important for cell functioning. Transport of these amino acids across intestinal cellular membranes occurs via L-type amino acid transporters. [68] These transporters belong to the family of the glycoprotein-associated amino acid transporters (gpaATs) and include LAT1, LAT2 y+LAT1, y+LAT2, and CT proteins, which are all linked by a disulfide bond to the same protein, i.e., 4F2hc. The 4F2hc protein is encoded by the glycoprotein solute carrier family 3 member gene SLC3A2.[69],[70] The y+LAT1 and 4F2hc together comprise an amino acid transporter, which belongs to the heterodimeric amino acid transporter (HAT) family. [68] This transporter releases intracellular dibasic amino acids in exchange for extracellular neutral amino acids and Na + . We have examined the significance of caveolae-dependent endocytosis in polyamine uptake using cell lines and GEM models with knockout of Cav-1 and Nos2 genes. [71] We found that knocking down of CAV-1 protein increases polyamine uptake in colon cancer-derived HCT116 cells. In Cav1−/− mice, the accumulated levels of dietarily-supplied putrescine in the colon and liver tissues were higher compared to the levels in wild-type littermates. In Nos2−/− mice, the accumulation of dietary putrescine in the colon and liver was abolished. DFMO treatment increased the small intestinal and colonic mucosal polyamine levels upon putrescine supplementation. Genetic suppression of the solute carrier transporter SLC3A2 in colon cancer cells reduced accumulation of exogenous putrescine and total polyamine content in DFMO-treated cells. [71] These experiments demonstrate the importance of SLC3A2 expression for polyamine transport under conditions of low tissue polyamine levels. To further validate this finding we evaluated the levels of the Slc3a2 RNA in both intestinal and colonic mucosa of 110-day-old Apc Min/+ mice (unpublished data). A significant (2.13-fold) upregulation of Slc3a2 transcript was observed in the small intestine of the ApcMin/+ mice compared to wild-type littermates. In the colon there was a four-fold decrease in the Slc3a2 transcript level in Apc mutant mice. These data are consistent with the interpretation that polyamine export, which is mediated by y+LAT1/4F2hc, is downregulated by loss of wild-type APC and can contribute to elevated colonic polyamines in APC-dependent colon carcinogenesis. The differential expression of this transporter may also contribute to the different effects of dietary arginine on intestinal and colonic tumorigenesis.
Conclusion
Cellular polyamine levels are regulated by genetic risk factors as well as dietary polyamine sources. Some disturbances in the control of polyamine homeostasis result in an increase of intracellular polyamine levels, which can promote tumorigenesis. A variety of GEM models have been generated to target the genes involved in the polyamine pathway. These models are extremely useful for investigating the functions of polyamine-related genes and associated pathologies as well as for testing potential preventive and therapeutic agents in preclinical studies. There still remains a need to develop animal models addressing the significance of the genetic variability within genes involved in polyamine metabolism; this is an important goal for future research.
Acknowledgments
The authors apologize to the many contributors to this field whose work has not been cited here due to space constraints. Our work in this area has been primarily supported by USPHS grants from the National Cancer Institute (CA-72008, CA-95060, and CA-123065). Drs Ignatenko and Besselsen declare that they have no competing interests. Dr Gerner has an ownership interest in Cancer Prevention Pharmaceuticals, Tucson, Arizona.[72]
References
| 1. | Igarashi K, Kashiwagi K. Polyamines: mysterious modulators of cellular functions. Biochem Biophys Res Commun 2000;271:559-64.  |
| 2. | Matsufuji S, Matsufuji T, Miyazaki Y, Murakami Y, Atkins JF, Gesteland RF, et al. Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell 1995;80:51-60. |
| 3. | Pegg AE. Mammalian polyamine metabolism and function. IUBMB Life 2009;61:880-94. |
| 4. | Schipper RG, Penning LC, Verhofstad AA. Involvement of polyamines in apoptosis. Facts and controversies: effectors or protectors? Semin Cancer Biol 2000;10:55-68. |
| 5. | Thomas T, Thomas TJ. Polyamine metabolism and cancer. J Cell Mol Med 2003;7:113-26. |
| 6. | Gerner EW, Meyskens FL Jr. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer 2004;4:781-92. |
| 7. | Persson L. Polyamine homoeostasis. Essays Biochem 2009;46:11-24. |
| 8. | Meyskens FL Jr, Gerner EW. Development of difluoromethylornithine as a chemoprevention agent. Clin Cancer Res 1999;5:945-51. |
| 9. | Meyskens FL Jr, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila) 2008;1:32-8. |
| 10. | Pendeville H, Carpino N, Marine JC, Takahashi Y, Muller M, Martial JA, et al. The ornithine decarboxylase gene is essential for cell survival during early murine development. Mol Cell Biol 2001;21:6549-58. |
| 11. | Kauppinen RA, Alhonen LI. Transgenic animals as models in the study of the neurobiological role of polyamines. Prog Neurobiol 1995;47:545-63. |
| 12. | Pegg AE. Regulation of ornithine decarboxylase. J Biol Chem 2006;281:14529. |
| 13. | Nilsson JA, Maclean KH, Keller UB, Pendeville H, Baudino TA, Cleveland JL. Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation. Mol Cell Biol 2004;24:1560-9. |
| 14. | Coffino P. Regulation of cellular polyamines by antizyme. Nat Rev Mol Cell Biol 2001;2:188-94. |
| 15. | Heller JS, Fong WF, Canellakis ES. Induction of a protein inhibitor to ornithine decarboxylase by the end products of its reaction. Proc Natl Acad Sci U S A 1976;73:1858-62. |
| 16. | Hoshino K, Momiyama E, Yoshida K, Nishimura K, Sakai S, Toida T, et al. Polyamine transport by mammalian cells and mitochondria: role of antizyme and glycosaminoglycans. J Biol Chem 2005;280:42801-8. |
| 17. | Mitchell JL, Judd GG, Bareyal-Leyser A, Ling SY. Feedback repression of polyamine transport is mediated by antizyme in mammalian tissue-culture cells. Biochem J 1994;299:19-22. |
| 18. | Feith DJ, Shantz LM, Pegg AE. Targeted antizyme expression in the skin of transgenic mice reduces tumor promoter induction of ornithine decarboxylase and decreases sensitivity to chemical carcinogenesis. Cancer Res 2001;61:6073-81. |
| 19. | Hayashi S, Murakami Y, Matsufuji S. Ornithine decarboxylase antizyme: a novel type of regulatory protein. Trends Biochem Sci 1996;21:27-30. |
| 20. | Wan T, Hu Y, Zhang W, Huang A, Yamamura K, Tang H. Changes in liver gene expression of Azin1 knock-out mice. Z Naturforsch C 2010;65:519-27. |
| 21. | Pegg AE, Xiong H, Feith DJ, Shantz LM. S-adenosylmethionine decarboxylase: structure, function and regulation by polyamines. Biochem Soc Trans 1998;26:580-6. |
| 22. | Kahana C. Ubiquitin dependent and independent protein degradation in the regulation of cellular polyamines. Amino Acids 2007;33:225-30. |
| 23. | Yerlikaya A, Stanley BA. S-adenosylmethionine decarboxylase degradation by the 26 S proteasome is accelerated by substrate-mediated transamination. J Biol Chem 2004;279:12469-78. |
| 24. | Nishimura K, Nakatsu F, Kashiwagi K, Ohno H, Saito T, Igarashi K. Essential role of S-adenosylmethionine decarboxylase in mouse embryonic development. Genes Cells 2002;7:41-7. |
| 25. | Heljasvaara R, Veress I, Halmekytö M, Alhonen L, Jänne J, Laajala P, et al. Transgenic mice overexpressing ornithine and S-adenosylmethionine decarboxylases maintain a physiological polyamine homoeostasis in their tissues. Biochem J 1997;323:457-62. |
| 26. | Forshell TP, Rimpi S, Nilsson JA. Chemoprevention of B-cell lymphomas by inhibition of the Myc target spermidine synthase. Cancer Prev Res (Phila) 2010;3:140-7. |
| 27. | Meyer RA Jr, Henley CM, Meyer MH, Morgan PL, McDonald AG, Mills C, et al. Partial deletion of both the spermine synthase gene and the Pex gene in the X-linked hypophosphatemic, gyro (Gy) mouse. Genomics 1998;48:289-95. |
| 28. | Ikeguchi Y, Wang X, McCloskey DE, Coleman CS, Nelson P, Hu G, et al. Characterization of transgenic mice with widespread overexpression of spermine synthase. Biochem J 2004;381:701-7. |
| 29. | Wang X, Levic S, Gratton MA, Doyle KJ, Yamoah EN, Pegg AE. Spermine synthase deficiency leads to deafness and a profound sensitivity to alpha-difluoromethylornithine. J Biol Chem 2009;284:930-7. |
| 30. | Pegg AE. Spermidine/spermine-N(1)-acetyltransferase: a key metabolic regulator. Am J Physiol Endocrinol Metab 2008;294:E995-1010. |
| 31. | Golab F, Kadkhodaee M, Zahmatkesh M, Hedayati M, Arab H, Schuster R, et al. Ischemic and non-ischemic acute kidney injury cause hepatic damage. Kidney Int 2009;75:783-92. |
| 32. | Wang Y, Casero RA Jr. Mammalian polyamine catabolism: a therapeutic target, a pathological problem, or both? J Biochem 2006;139:17-25. |
| 33. | Zahedi K, Lentsch AB, Okaya T, Barone S, Sakai N, Witte DP, et al. Spermidine/spermine-N1-acetyltransferase ablation protects against liver and kidney ischemia-reperfusion injury in mice. Am J Physiol Gastrointest Liver Physiol 2009;296:G899-909. |
| 34. | Wang Y, Devereux W, Stewart TM, Casero RA Jr. Cloning and characterization of human polyamine-modulated factor-1, a transcriptional cofactor that regulates the transcription of the spermidine/spermine N(1)-acetyltransferase gene. J Biol Chem 1999;274:22095-101. |
| 35. | Casero RA, Pegg AE. Polyamine catabolism and disease. Biochem J 2009;421:323-38. |
| 36. | Niiranen K, Pietilä M, Pirttilä TJ, Järvinen A, Halmekytö M, Korhonen VP, et al. Targeted disruption of spermidine/spermine N1-acetyltransferase gene in mouse embryonic stem cells. Effects on polyamine homeostasis and sensitivity to polyamine analogues. J Biol Chem 2002;277:25323-8. |
| 37. | Tucker JM, Murphy JT, Kisiel N, Diegelman P, Barbour KW, Davis C, et al. Potent modulation of intestinal tumorigenesis in Apcmin/+ mice by the polyamine catabolic enzyme spermidine/spermine N1-acetyltransferase. Cancer Res 2005;65:5390-8. |
| 38. | Alhonen L, Heikkinen S, Sinervirta R, Halmekytö M, Alakuijala P, Jänne J. Transgenic mice expressing the human ornithine decarboxylase gene under the control of mouse metallothionein I promoter. Biochem J 1996;314:405-8. |
| 39. | Pietilä M, Alhonen L, Halmekytö M, Kanter P, Jänne J, Porter CW. Activation of polyamine catabolism profoundly alters tissue polyamine pools and affects hair growth and female fertility in transgenic mice overexpressing spermidine/spermine N1-acetyltransferase. J Biol Chem 1997;272:18746-51. |
| 40. | Kee K, Foster BA, Merali S, Kramer DL, Hensen ML, Diegelman P, et al. Activated polyamine catabolism depletes acetyl-CoA pools and suppresses prostate tumor growth in TRAMP mice. J Biol Chem 2004;279:40076-83. |
| 41. | Min SH, Simmen RC, Alhonen L, Halmekyto M, Porter CW, Janne J, et al. Altered levels of growth-related and novel gene transcripts in reproductive and other tissues of female mice overexpressing spermidine/spermine N1-acetyltransferase (SSAT). J Biol Chem 2002;277:3647-57. |
| 42. | US Department of Agriculture. Agricultural Research Service. USDA National Nutrient Database for Standard Reference. Release 18, 2005 Nutrient Data Laboratory Home page. http://www.nal.usda.gov/fnic/foodcomp . |
| 43. | Zoumas-Morse C, Rock CL, Quintana EL, Neuhouser ML, Gerner EW, Meyskens FL Jr. Development of a polyamine database for assessing dietary intake. J Am Diet Assoc 2007;107:1024-7. |
| 44. | Deignan JL, Livesay JC, Shantz LM, Pegg AE, O′Brien WE, Iyer RK, et al. Polyamine homeostasis in arginase knockout mice. Am J Physiol Cell Physiol 2007;293:C1296-1301. |
| 45. | Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010;60:277-300. |
| 46. | Fodde R. The APC gene in colorectal cancer. Eur J Cancer 2002;38:867-71. |
| 47. | Gryfe R, Swallow C, Bapat B, Redston M, Gallinger S, Couture J. Molecular biology of colorectal cancer. Curr Probl Cancer 1997;21:233-300. |
| 48. | Bos JL. Ras oncogenes in human cancer: a review. Cancer Res 1989;49:4682-9. |
| 49. | Carson DA, Lois A. Cancer progression and p53. Lancet 1995;346:1009-11. |
| 50. | Chao A, Thun MJ, Connell CJ, McCullough ML, Jacobs EJ, Flanders WD, et al. Meat consumption and risk of colorectal cancer. JAMA 2005;293:172-82. |
| 51. | Murata M, Takayama K, Choi BC, Pak AW. A nested case-control study on alcohol drinking, tobacco smoking, and cancer. Cancer Detect Prev 1996;20:557-65. |
| 52. | Singh PN, Fraser GE. Dietary risk factors for colon cancer in a low-risk population. Am J Epidemiol 1998;148:761-74. |
| 53. | Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, et al. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell 1993;75:631-9. |
| 54. | Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990;247:322-4. |
| 55. | Bussey. Historical developments in familial adenomatous polypossis. In: Liss AR, editor. Familial Adenomatous Polyposis. New York, NY; 1990. p. 1-22 |
| 56. | Erdman SH, Ignatenko NA, Powell MB, Blohm-Mangone KA, Holubec H, Guillén-Rodriguez JM, et al. APC-dependent changes in expression of genes influencing polyamine metabolism, and consequences for gastrointestinal carcinogenesis, in the Min mouse. Carcinogenesis 1999;20:1709-13. |
| 57. | Fultz KE, Gerner EW. APC-dependent regulation of ornithine decarboxylase in human colon tumor cells. Mol Carcinog 2002;34:10-8. |
| 58. | Ignatenko NA, Holubec H, Besselsen DG, Blohm-Mangone KA, Padilla-Torres JL, Nagle RB, et al. Role of c-Myc in intestinal tumorigenesis of the ApcMin/+ mouse. Cancer Biol Ther 2006;5:1658-64. |
| 59. | Pietilä M, Parkkinen JJ, Alhonen L, Jänne J. Relation of skin polyamines to the hairless phenotype in transgenic mice overexpressing spermidine/spermine N-acetyltransferase. J Invest Dermatol 2001;116:801-5. |
| 60. | Berger FG, Kramer DL, Porter CW. Polyamine metabolism and tumorigenesis in the Apc(Min/+) mouse. Biochem Soc Trans 2007;35:336-9. |
| 61. | Yerushalmi HF, Besselsen DG, Ignatenko NA, Blohm-Mangone KA, Padilla-Torres JL, Stringer DE, et al. Role of polyamines in arginine-dependent colon carcinogenesis in Apc(Min) (/+) mice. Mol Carcinog 2006;45:764-73. |
| 62. | Yerushalmi HF, Besselsen DG, Ignatenko NA, Blohm-Mangone KA, Padilla-Torres JL, Stringer DE, et al. The role of NO synthases in arginine-dependent small intestinal and colonic carcinogenesis. Mol Carcinog 2006;45:93-105. |
| 63. | Zell JA, Ignatenko NA, Yerushalmi HF, Ziogas A, Besselsen DG, Gerner EW, et al. Risk and risk reduction involving arginine intake and meat consumption in colorectal tumorigenesis and survival. Int J Cancer 2007;120:459-68. |
| 64. | Löser C, Eisel A, Harms D, Fölsch UR. Dietary polyamines are essential luminal growth factors for small intestinal and colonic mucosal growth and development. Gut 1999;44:12-6. |
| 65. | Ignatenko NA, Besselsen DG, Roy UK, Stringer DE, Blohm-Mangone KA, Padilla-Torres JL, et al. Dietary putrescine reduces the intestinal anticarcinogenic activity of sulindac in a murine model of familial adenomatous polyposis. Nutr Cancer 2006;56:172-81. |
| 66. | Ignatenko NA, Besselsen DG, Stringer DE, Blohm-Mangone KA, Cui H, Gerner EW. Combination chemoprevention of intestinal carcinogenesis in a murine model of familial adenomatous polyposis. Nutr Cancer 2008;60:30-5. |
| 67. | Roy UK, Rial NS, Kachel KL, Gerner EW. Activated K-RAS increases polyamine uptake in human colon cancer cells through modulation of caveolar endocytosis. Mol Carcinog 2008;47:538-53. |
| 68. | Closs EI, Simon A, Vékony N, Rotmann A. Plasma membrane transporters for arginine. J Nutr 2004;134:2752S-29S; discussion 2765S-2767S. |
| 69. | Segawa H, Fukasawa Y, Miyamoto K, Takeda E, Endou H, Kanai Y. Identification and functional characterization of a Na+-independent neutral amino acid transporter with broad substrate selectivity. J Biol Chem 1999;274:19745-51. |
| 70. | Torrents D, Estévez R, Pineda M, Fernández E, Lloberas J, Shi YB, et al. Identification and characterization of a membrane protein (y+L amino acid transporter-1) that associates with 4F2hc to encode the amino acid transport activity y+L. A candidate gene for lysinuric protein intolerance. J Biol Chem 1998;273:32437-45. |
| 71. | Uemura T, Stringer DE, Blohm-Mangone KA, Gerner EW. Polyamine transport is mediated by both endocytic and solute carrier transport mechanisms in the gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol 2010;299:G517-522. |
| 72. | Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 2003;124:762-77. |
Authors
Natalia A. Ignatenko, Ph.D. is a Research Associate Professor of Cell Biology & Anatomy at the College of Medicine, University of Arizona, Tucson, Arizona. She is currently a Co-Director of the Experimental Mouse Shared Service at the Arizona Cancer Center.
Eugene W. Gerner, Ph.D. is a Professor Emeritus of Cell Biology & Anatomy and a Director of an NIH-funded Specialized Programs of Research Excellence (SPORE) in GI Cancer.
David G. Besselsen, D.V.M., Ph.D. is currently an Associate Director, University Animal Care, University of Arizona. He is a Diplomate, American College of Laboratory Animal Medicine and Diplomate, American College of Veterinary Pathology.
Figures
[Figure 1]
Tables