Bhawna Sharma, Rakesh K Singh
Department of Pathology and Microbiology, The University of Nebraska Medical Center, 985900 Nebraska Medical Center, Omaha, NE, 68198-5900, USA
| Date of Submission | 07-Jun-2011 |
| Date of Acceptance | 07-Oct-2011 |
| Date of Web Publication | 22-Dec-2011 |
Correspondence Address:
Rakesh K Singh
Department of Pathology and Microbiology, The University of Nebraska Medical Center, 985900 Nebraska Medical Center, Omaha, NE, 68198-5900
USA
Source of Support: None, Conflict of Interest: None
DOI: 10.4103/1477-3163.91119
Abstract
Therapy resistance is a major concern while treating breast cancer. Various mechanisms have been proposed, but so far nothing has been able to effectively address this problem. Accumulating evidences suggest that a subset of cancer cells provides survival benefits to the tumor and are responsible for therapy resistance and relapse of cancer. These so called the cancer stem cells, are known to be regulated by several pathways. Evidences shows that the tumor microenvironment plays a crucial role in maintaining the cancer stem cell pool. Signaling within the tumor is modulated by surrounding cells which secrete signals favoring tumor growth and metastasis. In breast cancer, the cancer stem cells have recently been reported to be influenced by tumor microenvironment via cytokines which act as chemoattractants for leukocytes. This review elucidates the emerging role of chemokine receptor and receptor activator of NFκB (RANK) ligand/RANK signaling pathways in mediating therapy resistance of breast cancer by maintaining the cancer stem cell pool.
Keywords: Breast cancer stem cells, chemokines, NFkB (RANK) ligand, therapy resistance
How to cite this article:
Sharma B, Singh RK. Emerging candidates in breast cancer stem cell maintenance, therapy resistance and relapse. J Carcinog 2011;10:36
How to cite this URL:
Sharma B, Singh RK. Emerging candidates in breast cancer stem cell maintenance, therapy resistance and relapse. J Carcinog [serial online] 2011 [cited 2021 Oct 14];10:36. Available from: https://carcinogenesis.com/text.asp?2011/10/1/36/91119
Introduction
Breast cancer is the most common cancer among females and accounts for more than 15% of the total deaths due to cancer each year. [1] It is not a single disease, but a set of diseases with no definitive cure. [2] Although many treatments have been developed for breast cancer, still a gap in knowledge exists. The ability to target malignant stages would facilitate more effective treatment. [3],[4],[5] Therapy resistance represents a significant hurdle in the treatment of breast cancer forcing the development of alternative strategies. Accumulation of genetic and epigenetic changes in the normal cells has been suggested to be the leading cause of tumor intiation but the exact cause of cancer is unknown. It has been shown that a hierarchy exists in breast cancer with some cells having the ability to divide and proliferate extensively while others only have limited proliferative ability in vivo. [6] This heterogeneity raises the question about which cells to target. The emergence of cancer stem cell hypothesis has prompted question that have attempted to unravel the importance of stem cells in cancer. According to cancer stem cell hypothesis, cancer stem cells (CSCs) are the transformed stem cells or progenitor cells with acquired self-renewal properties of stem cells and are responsible for tumor proliferation, metastasis, tumor relapse and moreover, resistance to chemo- and radiation therapy. [7],[8],[9] Various mechanisms have been implicated for therapy resistance, but the exact mechanism leading to the selection of CSCs for resistance in breast cancer has not been elucidated well.
The biology of resistance in breast cancer is complex and is getting more intricate with the discovery of CSCs. [10] Many pathways have been studied in relation to resistance, but so far nothing has been able to resolve this issue. Multiple investigators have implicated breast cancer cells with stem cell like properties to be involved in resistance. [6],[11],[12],[13] Breast cancer cells with stem cell like properties can be defined by the expression of surface markers: ESA + , CD44 + , CD24−/low Lin− similar to the hematopoietic stem cells. [14],[15],[16],[17],[18] How breast CSC evades chemotherapy still needs to be explored and is the subject of ongoing research. This review focuses on the emerging role of cytokines in facilitating therapy resistance by maintaining the breast CSC pool. Cytokines, specifically chemokines, has been cited as important mediators of tumorigenesis in various cancers and their signaling have been modulated in several ways to circumvent cancer. In addition, RANK/RANKL signaling has recently been proposed to be an important component in mammary carcinogenesis, specifically in the maintenance of breast cancer stem cells. [19],[20] The review mainly concentrates on the key functions of chemokines and RANK/RANKL signaling in breast cancer and how they provide survival benefits to breast cancer cells after chemotherapy.
Chemokine and Breast Cancer
Inflammation and inflammatory cytokines are critical contributor to tumor progression. Chemokines are the group of cytokines which were originally identified as chemoattractants for leukocytes. They can be divided into various sub-groups based on the cysteine residue present on their amino terminus. Chemokines are secreted proteins that regulate cell behavior via G-protein coupled receptors, and orchestrate tissue inflammation by recruitment and activation of leukocytes. Constitutive expression of pro-inflammatory chemokines, a hallmark of many human cancers, helps establish a supportive tumor stroma, and in some cases, directly stimulates tumor proliferation and invasion via receptors on the tumor cells. [21]
The tumor stroma plays a crucial role in supporting the growth of cancer cells. Within a tumor, surrounding and infiltrating cells secrete factors that favor tumor growth and metastasis and regulate signaling events. In the case of tumors, these cell include fibroblasts, inflammatory cells, endothelial cells and mesenchymal stem cells. [22] The microenvironment or niche not only plays a role in malignant cell survival but also in normal organ development. Microenvironment, or niche helps stem cells, maintains their stemness and prevents their differentiation into different cell types. [23],[24] Interactions between cancer stem cells and their microenvironment alter the self-renewal pathways and cytokine loop, which helps tumor cells modulate the inflammatory response for their benefit. Several studies have suggested designing therapies to target self-renewal pathways, which are considered key operating events in CSCs. [25],[26],[27],[28],[29] However, targeting self-renewal pathways to eradicate CSCs can be toxic as these pathways are also used by normal stem cells. Chronic inflammation drives tumorigenesis, and tumors are inherently pro-inflammatory with infiltrating leukocytes which are thought to be critical for their maintenance and progression. Thus, molecules driving tumor-associated inflammation have considerable potential as therapeutic targets and fewer chances of systematic toxicities to the normal tissue. Chemokines along with their receptor have been shown to play an important role in the progression of cancer and metastasis. CXCL12, through autocrine signaling, has been reported to give rise to carcinoma-associated fibroblasts which are activated fibroblasts in the tumor microenvironment, and have been shown to substantially promote tumorigenesis in breast cancer. [30] CXCL12, after binding to its receptor CXCR4, has been shown to enhance proliferation of breast cancer cells and stimulate angiogenesis. [31] Chemokines are potent attractants for leukocytes, which are useful in clearing the tumor. However, a recent report suggests that when CXCL12 was over-expressed it was found to inhibit metastasis and tumor growth in a syngenic mouse model of breast carcinoma. [32] This finding emphasizes that exploitation of any signaling pathways should be done carefully to design effective therapy.
CXCR2 and its ligands, CXCL1 and CXCL5, have recently been reported to promote migration of polyoma virus middle-T (PyMT) mammary cancer cell lines (derived from the polyoma PyMT mice), when these cells were treated with conditioned media derived from mesenchymal stem cells. These findings reinforce the fact that cytokines are important mediators in breast cancer. [33] Higher expression of CXCL8 and other CXCR2 ligand has been observed in the serum of patients with breast carcinomas and has been related to poor outcome in these patients. [34],[35]
Chemokines direct trafficking of leukocytes into tumors, promote angiogenesis by attracting leukocytes, and interact with endothelial cells. Recently CCL18, secreted by tumor-associated macrophages, has been reported to promote malignancy and metastasis in breast cancer. Specifically, CCL18 has been shown to trigger clustering of intergrins along with enhancing their adherence to the extracellular matrix promoting the invasiveness of breast cancer cells. [36]
Role of Chemokine in the Maintenance of Breast Cancer Stem Cells
CSCs are a subset of cancer cell population implicated in initiating and maintaining tumor cells. CD44 + cells have been suggested to be breast cancer stem cells since, CD44 + CD24 -/low Lineage – cells derived from malignant pleural effusions of breast cancer patients show increased ability to proliferate and recapitulate the heterogeneous population of a primary tumor in in vivo xenograft models of NOD/SCID mice. [6] Levels of CXCL8 have been found to be elevated in breast cancer cell lines with higher frequency of CD44 + /CD24− cells, suggesting this chemokine should be considered as a marker for the identification of breast CSCs. [37] ALDEFLUOR flow cytometry is an assay used to characterize breast CSCs. Gene expression profiling of 23 aldehyde dehydrogenase (ALDH1) positive breast cancer cell lines identified CXCL8 and its receptor, CXCR1, as important genes that are upregulated in CSCs. Furthermore, treatment of ALDH1 positive cells with recombinant CXCL8 promotes mammosphere (in vitro assay for stemness) formation in these cells, confirming the fact that CXCL8 mediated CXCR1 signaling plays an important role in sustaining the CSC population. [38]
Blocking of CXCR1 in breast cancer cell lines using small molecular inhibitor, repertaxin, or CXCR1-specific antibodies showed reduction in the CSC population in vitro. Targeting CXCR1 signaling also induced tumors cell apoptosis, retarded tumor growth and lowered metastasis [39] In addition, chemotherapy increases CXCL8 production by breast cancer cells and the emerging relationship of this chemokine with CSCs suggests that CXCL8 may contribute to the enhancement of the CSC pool after chemotherapy. [25]
Recently, a cytokine loop formed by CXCL7 and IL6 has been found to be important in regulating the breast CSC population by bone marrow derived mesenchymal stem cells. A study by Liu,S et al,[40] provides evidence that mesenchymal stem cell might be directing self-renewal in CSC population via chemokines in the tumor microenvironment. Accumulating literature about the involvement of chemokine in maintaining CSC pool suggests that targeting chemokine-mediated pathways may provide an opportunity to eradicate this subset of cancer cells.
Therapy Resistance and Cancer Stem Cells in Breast Cancer
Investigational cancer therapy has struggled to perform in clinical drug development, with three times lower rate of success than cardiovascular disease. [41] As mentioned earlier, despite the development of new strategies the incidence of and from death due breast cancer is high, mainly because of chemotherapy-resistance. Studies involving breast CSCs show that cells expressing CSC markers are resistant to radiation. Resistance has been seen in both in vitro and in vivo models of breast cancer stem cell response to chemo or radiation therapy. [42],[43] This resistance is further increased after ectopic expression of Wnt or β-catenin which are involved in developmental pathways and self-renewal of normal stem cells. [44],[45] These studies suggest that therapy activates the pathways involved in self-renewal of normal stem cells in cancer cells or increases the quiescent stem cell population or triggers dying cells to send signals to cancer stem cells to proliferate. The CSCs, like normal stem cells, are hypothesized to undergo asymmetric cell division giving rise to one progenitor for self-renewal and a differentiated cancer cell. Studies suggest that breast tumors may be initiated and maintained by these cells which possess stem cell-like characteristics. [46] These tumor cells are resistant to therapy and are able to metastasize to distant organs. Furthermore, the emergence of cells expressing stem cell markers after neo-adjuvant therapy in cancer patients and in clinical trials also supports the in vitro and in vivo findings. The above findings emphasize an urgent need to target CSCs to win a battle against breast cancer.
Chemokine and Breast Cancer Therapy Resistance
Chemokines have been shown to play an important role in tumor growth and progression. Studies on breast cancer suggested that levels of CXCL1, CXCL6 and CXCL8 increase after chemotherapy, [47],[48],[49],[50] which may be responsible for chemoresistance, aid in the survival of tumor cells, and increases the CSC population. Reports have shown that in in vitro setting, cancer cells which survive the initial phase of chemotherapy express higher level of CXCL8. [47],[48] Expression of CXCL8 by malignant cells has been suggested to be one of the escape mechanisms used by cancer cells to evade cell death. The exact mechanism by which CXCL8 signaling provides survival benefits to the cancer cells needs to be elucidated. Similarly, CXCL12/CXCR4 chemokine receptor axis provides resistance against anti-estrogen therapy in CXCR4-driven tumors when treated with exogenous CXCL12. [51] These studies indicate that chemokines provide survival benefits to breast cancer cells.
Rank-Rankl Signaling and Breast Cancer
TNF-α an inflammatory cytokine shown to support tumor growth, highly expressed in breast carcinomas. Infact, detection of higher numbers of cells expressing TNF-α in breast tumors has been found to be associated with increasing tumor grade and node involvement. [52],[53] Furthermore, breast tumors as well as breast cancer cell lines have been reported to express RANK and RANKL, a cytokine in TNF-α superfamily. However, their expression varies among different types of breast tumors. [54],[55] Data from immunohistochemical studies on breast tumor tissues revealed that 90% of the normal breast tissue, 60% of the non-metastatic invasive ductal carcinoma (IDC), 31% of metastatic IDC, and 2% of osteolytic bone tumor metastases (BTM) lesions expresses RANK. [56] Similar studies from another group revealed that with increasing histological grade the expression of RANKL decreases. However, they also observed that the tumors which retain the RANKL expression are estrogen receptor negative (ER− ) and are of high histological grade. [57] A recent study reported higher expression of RANKL in IDCs than in ductal carcinomas in situ (DCIS) or lymph-node-positive tumors. [58] These studies together propose that there is a loss of RANKL in breast tumors as they become more aggressive, but the one that retain RANKL are of higher histological grade. RANK-RANKL signaling has been documented to play an important role in bone metastasis of breast cancer. Breast cancer cells secrete parathyroid hormone related peptide (PTHrP) which stimulates production of RANKL by osteoblasts which in turn activates osteoclasts leads to osteolysis [59] in breast cancer patients.
Rank-Rankl and Breast Cancer Therapy Resistance
Mammary epithelial cells and most of the breast cancer cell lines express RANK. [54],[55],[56],[57] Recently, it has been observed that RANKL protects murine mammary epithelial and human SKBR3 breast cancer cells from apoptosis following treatment with 1 μM of doxorubicin in the presence of 1 μg/ml of RANKL. [20] Doxorubicin treatment in the presence of RANKL results in a higher percentage of SKBR3 cells in M3 (S/G2/M-phase) when compared to only doxorubicin treated cells. Furthermore, the more aggressive breast cancer cell lines, like MDA-MB231 have also been shown to express RANK at the mRNA level [55] implicating that these cells will also respond to RANKL stimulus in a similar fashion as SKBR3 cells. These data collectively suggest the role of RANK-RANKL signaling in mediating chemotherapy resistance in breast cancer cells.
Similarly, recent report shows that treatment of murine mammary epithelial cells (MECs) and human SKBR3 breast cancer cells with exogenous RANKL protects them from γ-irradiation induced cell death. [20] Flow cytometric analysis of the MECs and SKBR3 cells in the presence of RANKL after treatment with γ-irradiation showed fewer cells in the sub-G1(M1) phase in comparison to treatment with γ-irradiation only. Moreover, in the presence of RANKL, up-regulation of γ-irradiation-induced pro-apoptotic molecules, Bim, Puma and Noxa, was not observed, suggesting that administration of RANKL down-regulates apoptotic pathways in these cells.
Nuclear factor (NF)κB has been demonstrated to influence various survival pathways in cancer cells and has been shown to be a downstream target of RANK/RANKL signaling during osteoclastogenesis. [60] In MECs, RANK/RANKL has been demonstrated to work through IKK -α (Inhibitor of nuclear factor kappa-B kinase subunit alpha)-NFκB-cyclin. [20] Similar mechanisms have been found to operate in human SKBR3 breast cancer cells where it was observed that after RANKL stimulation, expression of NFκB, p38 mitogen activated protein kinases (MAPKs) and extracellular signal regulated kinase (ERK) activation was induced. Further, RANKL was found to induce proliferation in SKBR3 cells when growth curve was analyzed in stimulated versus unstimulated cells. [20] These observations implicate RANK/RANKL as a regulator of breast cancer cell growth in presence of chemotherapeutic agents via NFκB pathway leading to resistance.
Rank-Rankl in Mammary Carcinogenesis and Cancer Stem Cell Maintenance
Recent studies have demonstrated the role of RANK-RANKL signaling in murine mammary stem cell (MaSC) maintenance. [61],[62] RANK-RANKL axis has been shown to be a major player in maintaining the MaSC pool in the mammary glands of mice. [61] These studies also illustrated that treatment of mice with progesterone enhanced the expression of RANK in MaSC, whereas similar treatment increased expression of RANKL in the luminal cells of mammary glands. Treatment of MaSC-enriched population and luminal cell population with RANKL-Fc inhibited clonogenic activity of the MaSC-enriched but not the luminal cell subset of cells. These findings suggest that RANK-RANKL signaling might be responsible for the increase of MaSC pool during pregnancy. There is a possibility that in a heterogenous tumor, normal stem cells which are more basal-like express RANK and luminal cells or normal mammary epithelium cells express RANKL and the paracrine signaling triggered by chemotherapeutic drugs enhances the survival of cancer stem cells.
Progesterone-driven mammary tumors have recently been linked with RANK-RANKL signaling in mammary gland. The effect of RANKL on tumor formation was studied in MPA/DMBA treated mice. Administration of RANK-Fc, concurrently with a carcinogen (DMBA), almost completely blocked the formation of mammary tumor in wild-type mice. Similar treatment with bisposphonate, used to inhibit osteoclastogenesis, instead of RANK-Fc, had no effect on tumor formation. [19] These experiments suggest a role for RANKL in mammary tumor formation independent of osteoclastogenesis mediated by it in bone microenvironment during cancer metastasis and arthritis. Moreover, RANK/RANKL expression has also been observed in the stroma as well as tumor epithelium of human breast cancer patients. [63],[64] These observations indicate mechanisms other than hormonal signaling may activate or dysregulate this pathway in breast cancer. Recent reports suggest the expression of RANK by MaSCs. The role of RANK with respect to tumor initiating cells (TIC) was tested by isolating cancer cells from MPA/DMBA treated wild type and RANK∆mam female mice. The freshly isolated cancer cells were tested for their ability to form primary mammospheres. After single cell separation, cells were when evaluated for the formation of secondary mammospheres; it was found that TIC from RANK∆mam mice was significantly impaired in their ability to form mammospheres. This indicates that loss of RANK expression significantly affects the self renewal capacity of putative cancer stem cells. [20] Similarly, SKBR3, human breast cancer cells when stimulated with RANKL have been shown to exhibit anchorage independent growth in a soft-agar colony formation assay. SKBR3 are able to form macroscopic colonies after 18 days when stimulated with RANKL in culture. This phenomenon is reversed by treatment with OPG, a decoy receptor for RANKL. [20] As formation of colonies on soft-agar is a characteristic of stem cells and correlate with tumorigenicity in vivo,[65] these findings imply that RANK-RANKL signaling could be responsible for favoring the stem cell population in breast tumors. All these results indicate that RANK-RANKL signaling plays a role in maintaining the breast cancer stem cell pool in breast tumors.
The Vicious Cycle Involving Chemokine and Rank-Rankl Signaling in the Maintenance of Breast Cancer Stem Cells and Chemotherapy Resistance
It has been demonstrated that malignant tumors which express RANKL are associated with poor prognosis and more aggressive disease. [56],[57] RANK/RANKL has been shown to have NFκB as one of its main downstream targets. [20] NFκB itself has been implicated in cancers and in driving immune responses. Constitutive expression/activation of NFκB has been observed in ER-negative and poorly differentiated primary breast tumors. [66] Stimulation with RANKL has been shown to promote anchorage-independent growth in human SKBR3 breast cancer cells, as mention earlier. The first line of treatment for breast cancer is chemotherapy followed by surgery. Aggressive breast tumors express RANK/RANKL. Up-regulation of NFκB by chemotherapeutic agents in cancer cells [67],[68] and the failure of TICs from RANK∆mam mice treated with MPA/ DMBA to form secondary mammosphere [19] suggest NFκB as a link between chemotherapy and RANK-RANKL signaling. Moreover, a recent study has shown that continuous selection with a chemotherapeutic agents selects cancer cells with stem cell like characteristics. [69] Based on the cumulative evidences this implicates that RANK/RANKL mediates the effect of chemotherapy by activating NFκB in breast CSCs. It can therefore be suggested that chemotherapeutic agents select resistant cells by triggering RANK-RANKL signaling leading to up-regulation of anti-apoptotic pathways via NFκB, providing survival benefits to breast tumors. Additional reports suggest a complex cooperation between NFκB and CXCL8 in invasive breast cancer cells. [70],[71],[72] Chemokines like CXCL8, CXCL1 are up-regulated after chemotherapy and chemoattract neutrophils. Neutrophils are known to express proteases. RANKL is a membrane-bound ligand and requires proteolytic release to act in a paracrine fashion. [73] There is a possibility that in tumors after chemotherapy up-regulation of CXCL8 up-regulate RANKL in breast cancer cells (through NFκB) as well as recruit neutrophils to the tumor site. Then neutrophils, through their proteases, cause cleavage of RANKL, releasing soluble RANKL (sRANKL) which binds to its receptor RANK on CSCs leading to their maintenance [Figure 1].
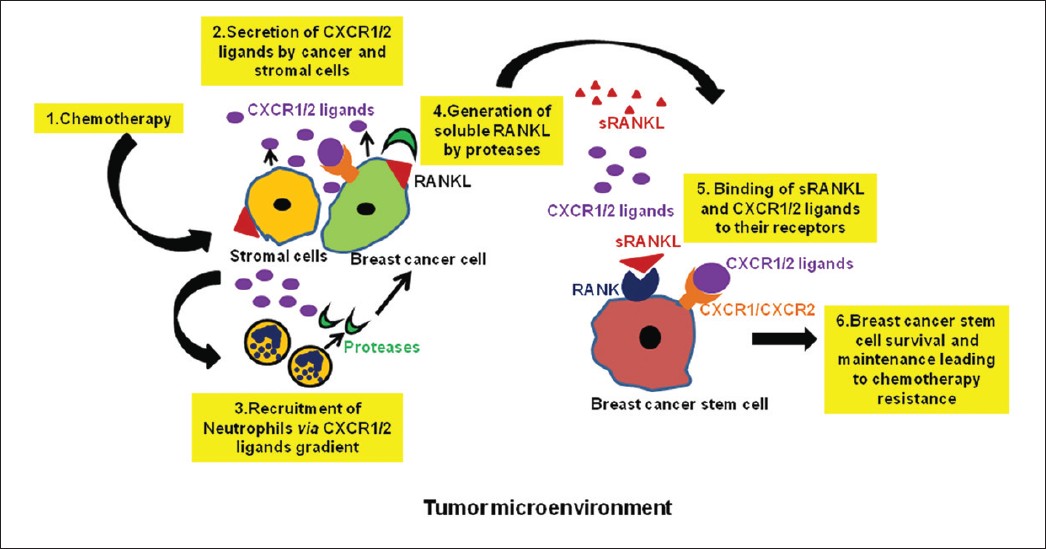 |
Figure 1: Vicious cycle involving chemokine and RANK-RANKL signaling in the maintenance of breast cancer stem cells and chemotherapy resistance: 1) In the tumor microenvironment, upon chemotherapeutic treatment, stromal and cancer cells 2) release CXCR1/2 ligands which help in the 3) recruitment of neutrophils to the cancer site. CXCR1/2 ligands can also bind to their receptor, CXCR1/CXCR2, expressed by cancer cell or cancer stem cells providing survival benefits to them. 4) Neutrophils, through their proteases, cleave RANKL present on cancer cells or stromal cells, leading to the generation of soluble RANKL (sRANKL). 5) Released sRANKL binds to its receptor, RANK, present on cancer stem cells, leading to their 6) survival and maintenance in the presence of chemotherapy Click here to view |
Conclusion
Chemokines, important in inflammation and tumor-associated inflammation, are emerging as interesting players in mediating therapy resistance in breast cancer through, maintenance of the CSC pool. It has been shown that levels of CXCL1, and CXCL8 increase in breast cancer cells after chemotherapy. In addition, CXCL8, IL6 and RANKL have been implicated in regulating breast CSC maintenance. Moreover, these cytokines have NFκB as one of their downstream targets, which is activated by chemotherapeutic agents, suggesting that cytokines release, in response to chemotherapy, maintain the CSC pool via RANK/RANKL signaling. CXCL8 has been observed to be over-expressed in breast cancer patients as well as in aggressive breast cancer cell lines. Furthermore, CXCL8 has been shown to up-regulate RANKL both at mRNA and protein levels in osteoblastic cells which leads to the activation of bone resorbing osteoclasts, suggesting that CXCL8 up-regulated by chemotherapy can utilize the same mechanism in primary tumor to maintain the CSC pool. Targeting chemokines, which are emerging as important factors involved in regulating breast CSCs, may provide promising therapeutic options to prevent therapy resistance and relapse in breast cancer patients.
Acknowledgments
This work was supported in part by Susan G. Komen for the Cure grant KG090860 COBRE grant RR021937 (Nebraska Center for Nanomedicine), and Cancer Center Support Grant (P30CA036727) from National Cancer Institute, National Institutes of Health and Nebraska Research Initiative Cancer Glycobiology Program (R.K.S.). Bhawna Sharma is supported through University of Nebraska Medical Center Graduate Student Fellowship/Assistantship.
References
| 1. | Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69-90.  |
| 2. | Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009;462:1005-10. |
| 3. | Nabholtz JM, Reese DM, Lindsay MA, Riva A. Combination chemotherapy for metastatic breast cancer. Expert Rev Anticancer Ther 2002;2:169-80. |
| 4. | Giaccone G, Pinedo HM. Drug Resistance. Oncologist 1996;1:82-7. |
| 5. | Rivera E, Gomez H. Chemotherapy resistance in metastatic breast cancer: the evolving role of ixabepilone. Breast Cancer Res 2010;12 Suppl 2:S2. |
| 6. | Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003;100:3983-8. |
| 7. | Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol 2005;45:872-7. |
| 8. | Fiala S. The cancer cell as a stem cell unable to differentiate. A theory of carcinogenesis. Neoplasma 1968;15:607-22. |
| 9. | Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105-11. |
| 10. | Dean M. Cancer stem cells: Implications for cancer causation and therapy resistance. Discov Med. 2005;5:278-82. |
| 11. | Dave B, Chang J. Treatment resistance in stem cells and breast cancer. J Mammary Gland Biol Neoplasia 2009;14:79-82. |
| 12. | Hambardzumyan D, Squatrito M, Holland EC. Radiation resistance and stem-like cells in brain tumors. Cancer Cell 2006;10:454-6. |
| 13. | Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res 2008;68:3243-50. |
| 14. | Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell 2006;124:1111-5. |
| 15. | Lynch MD, Cariati M, Purushotham AD. Breast cancer, stem cells and prospects for therapy. Breast Cancer Res 2006;8:211. |
| 16. | Polyak K, Hahn WC. Roots and stems: stem cells in cancer. Nat Med 2006;12:296-300. |
| 17. | Weissman I. Stem cell research: paths to cancer therapies and regenerative medicine. JAMA 2005;294:1359-66. |
| 18. | Wicha MS. Identification of murine mammary stem cells: Implications for studies of mammary development and carcinogenesis. Breast Cancer Res 2006;8:10. |
| 19. | Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010;468:103-7. |
| 20. | Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010;468:98-102. |
| 21. | Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett 2008;267:226-44. |
| 22. | Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer 2007;7:139-47. |
| 23. | Wicha MS. Cancer stem cells and metastasis: lethal seeds. Clin Cancer Res 2006;12:5606-7. |
| 24. | Spradling A, Drummond-Barbosa D, Kai T. Stem cells find their niche. Nature 2001;414:98-104. |
| 25. | Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol 2009;7:e1000121. |
| 26. | Mariani G, Fasolo A, De Benedictis E, Gianni L. Trastuzumab as adjuvant systemic therapy for HER2-positive breast cancer. Nat Clin Pract Oncol 2009;6:93-104. |
| 27. | Magnifico A, Albano L, Campaner S, Delia D, Castiglioni F, Gasparini P, et al. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin Cancer Res 2009;15:2010-21. |
| 28. | Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164-72. |
| 29. | Korkaya H, Wicha MS. HER-2, notch, and breast cancer stem cells: targeting an axis of evil. Clin Cancer Res 2009;15:1845-7. |
| 30. | Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A 2010;107:20009-14. |
| 31. | Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005;121:335-48. |
| 32. | Williams SA, Harata-Lee Y, Comerford I, Anderson RL, Smyth MJ, McColl SR. Multiple functions of CXCL12 in a syngeneic model of breast cancer. Mol Cancer 2010;9:250. |
| 33. | Halpern JL, Kilbarger A, Lynch CC. Mesenchymal stem cells promote mammary cancer cell migration in vitro via the CXCR2 receptor. Cancer Lett 2011;308:91-9. |
| 34. | Koz³owski L, Zakrzewska I, Tokajuk P, Wojtukiewicz MZ. Concentration of interleukin-6 (IL-6), interleukin-8 (IL-8) and interleukin-10 (IL-10) in blood serum of breast cancer patients. Rocz Akad Med Bialymst 2003;48:82-4. |
| 35. | Benoy IH, Salgado R, Van Dam P, Geboers K, Van Marck E, Scharpé S, et al. Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin Cancer Res 2004;10:7157-62. |
| 36. | Chen J, Yao Y, Gong C, Yu F, Su S, Chen J, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 2011;19:541-55. |
| 37. | Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, et al. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res 2006;8:R59. |
| 38. | Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res 2010;16:45-55. |
| 39. | Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120:485-97. |
| 40. | Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res 2011;71:614-24. |
| 41. | Kamb A. What’s wrong with our cancer models? Nat Rev Drug Discov 2005;4:161-5. |
| 42. | Hambardzumyan D, Squatrito M, Holland EC. Radiation resistance and stem-like cells in brain tumors. Cancer Cell 2006;10:454-6 |
| 43. | Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res 2008;68:3243-50. |
| 44. | Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst 2006;98:1777-85. |
| 45. | Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A 2007;104:618-23. |
| 46. | Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. Bio Drugs 2007;21:299-310. |
| 47. | De Larco JE, Wuertz BR, Rosner KA, Erickson SA, Gamache DE, Manivel JC, et al. A potential role for interleukin-8 in the metastatic phenotype of breast carcinoma cells. Am J Pathol 2001;158:639-46. |
| 48. | De Larco JE, Wuertz BR, Manivel JC, Furcht LT. Progression and enhancement of metastatic potential after exposure of tumor cells to chemotherapeutic agents. Cancer Res. 2001;61:2857-61. |
| 49. | Lev DC, Ruiz M, Mills L, McGary EC, Price JE, Bar-Eli M. Dacarbazine causes transcriptional up-regulation of interleukin 8 and vascular endothelial growth factor in melanoma cells: a possible escape mechanism from chemotherapy. Mol Cancer Ther 2003;2:753-63. |
| 50. | Maxwell PJ, Gallagher R, Seaton A, Wilson C, Scullin P, Pettigrew J, et al. HIF-1 and NF-kappaB-mediated upregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic prostate cancer cells. Oncogene 2007;26:7333-45. |
| 51. | Rhodes LV, Short SP, Neel NF, Salvo VA, Zhu Y, Elliott S, et al. Cytokine receptor CXCR4 mediates estrogen-independent tumorigenesis, metastasis, and resistance to endocrine therapy in human breast cancer. Cancer Res. 2011;71:603-13. |
| 52. | Leek RD, Landers R, Fox SB, Ng F, Harris AL, Lewis CE. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br J Cancer 1998;77:2246-51. |
| 53. | Miles DW, Happerfield LC, Naylor MS, Bobrow LG, Rubens RD, Balkwill FR, et al. Expression of tumour necrosis factor (TNF alpha) and its receptors in benign and malignant breast tissue. Int J Cancer 1994;56:777-82. |
| 54. | Rachner TD, Schoppet M, Niebergall U, Hofbauer LC. 17beta-Estradiol inhibits osteoprotegerin production by the estrogen receptor-alpha-positive human breast cancer cell line MCF-7. Biochem Biophys Res Commun. 2008;368:736-41. |
| 55. | Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006;440:692-6. |
| 56. | Bhatia P, Sanders MM, Hansen MF. Expression of receptor activator of nuclear factor-kappaB is inversely correlated with metastatic phenotype in breast carcinoma. Clin Cancer Res 2005;11:162-5. |
| 57. | Cross SS, Harrison RF, Balasubramanian SP, Lippitt JM, Evans CA, Reed MW, et al. Expression of receptor activator of nuclear factor kappabeta ligand (RANKL) and tumour necrosis factor related, apoptosis inducing ligand (TRAIL) in breast cancer, and their relations with osteoprotegerin, oestrogen receptor, and clinicopathological variables. J Clin Pathol 2006;59:716-20. |
| 58. | Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011;470:548-53. |
| 59. | Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002;2:584-93. |
| 60. | Jimi E, Aoki K, Saito H, D’Acquisto F, May MJ, Nakamura I, et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med 2004;10:617-24. |
| 61. | Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature 2010;465:798-802. |
| 62. | Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature 2010;465:803-7. |
| 63. | Mancino AT, Klimberg VS, Yamamoto M, Manolagas SC, Abe E. Breast cancer increases osteoclastogenesis by secreting M-CSF and upregulating RANKL in stromal cells. J Surg Res 2001;100:18-24. |
| 64. | Quinn JM, Elliott J, Gillespie MT, Martin TJ. A combination of osteoclast differentiation factor and macrophage-colony stimulating factor is sufficient for both human and mouse osteoclast formation in vitro. Endocrinology 1998;139:4424-7. |
| 65. | Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell 1974;3:355-9. |
| 66. | Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ Jr, Sledge GW Jr. Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol 1997;17:3629-39. |
| 67. | Brea-Calvo G, Siendones E, Sánchez-Alcázar JA, de Cabo R, Navas P. Cell survival from chemotherapy depends on NF-kappaB transcriptional up-regulation of coenzyme Q biosynthesis. PLoS One 2009;4:e5301. |
| 68. | Huang TT, Wuerzberger-Davis SM, Seufzer BJ, Shumway SD, Kurama T, Boothman DA, et al. NF-kappaB activation by camptothecin. A linkage between nuclear DNA damage and cytoplasmic signaling events. J Biol Chem 2000;275:9501-9. |
| 69. | Calcagno AM, Salcido CD, Gillet JP, Wu CP, Fostel JM, Mumau MD, et al. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst 2010;102:1637-52. |
| 70. | Kawada K, Sonoshita M, Sakashita H, Takabayashi A, Yamaoka Y, Manabe T, et al. Pivotal role of CXCR3 in melanoma cell metastasis to lymph nodes. Cancer Res 2004;64:4010-7. |
| 71. | De Larco JE, Wuertz BR, Rosner KA, Erickson SA, Gamache DE, Manivel JC, et al. A potential role for interleukin-8 in the metastatic phenotype of breast carcinoma cells. Am J Pathol 2001;158:639-46. |
| 72. | Xie JH, Nomura N, Lu M, Chen SL, Koch GE, Weng Y, et al. Antibody-mediated blockade of the CXCR3 chemokine receptor results in diminished recruitment of T helper 1 cells into sites of inflammation. J Leukoc Biol 2003;73:771-80. |
| 73. | Wilson TJ, Nannuru KC, Futakuchi M, Sadanandam A, Singh RK. Cathepsin G enhances mammary tumor-induced osteolysis by generating soluble receptor activator of nuclear factor-kappaB ligand. Cancer Res 2008;68:5803-11. |
Authors
Dr. Rakesh Singh, Department of Pathology and Microbiology, University of Nebraska Medical Center, 985900 Nebraska Medical Center, Omaha, NE
Ms. Bhawna Sharma, Department of Pathology and Microbiology, University of Nebraska Medical Center, 985900 Nebraska Medical Center, Omaha, NE
Figures
[Figure 1]