Mariangela De Robertis1, Emanuela Massi1, Maria Luana Poeta2, Simone Carotti3, Sergio Morini3, Loredana Cecchetelli4, Emanuela Signori5, Vito Michele Fazio6
1 Laboratory of Molecular Medicine and Biotechnology, CIR, Campus Bio-Medico University of Rome, Via Álvaro del Portillo 21 – 00128 Rome, Italy
2 Department of General and Environmental Physiology and Center of Excellence in Comparative Genomics (CEGBA), University of Bari, 70126, Bari, Italy
3 Department of Biomedical Research (CIR), University Campus Bio-Medico of Rome, Via Álvaro del Portillo 21 – 00128 Rome, Italy
4 IBI Istituto Biochimico Italiano Giovanni Lorenzini, Via Fossignano, 2 – 04011 Aprilia (LT), Italy
5 Institute of Translational Pharmacology. Laboratory of Molecular Pathology and Experimental Oncology. National Research Council (CNR), Via Fosso del Cavaliere 100, 00133 Rome, Italy
6 Laboratory of Molecular Medicine and Biotechnology, CIR, Campus Bio-Medico University of Rome, Via Álvaro del Portillo 21 – 00128 Rome; Laboratory of Oncology, IRCCS ‘Casa Sollievo della Sofferenza,’ Viale Padre Pio, 71013 San Giovanni Rotondo (FG), Italy
| Date of Submission | 14-Jan-2011 |
| Date of Acceptance | 05-Feb-2011 |
| Date of Web Publication | 24-Mar-2011 |
Correspondence Address:
Vito Michele Fazio
Laboratory of Molecular Medicine and Biotechnology, CIR, Campus Bio-Medico University of Rome, Via Álvaro del Portillo 21 – 00128 Rome; Laboratory of Oncology, IRCCS ‘Casa Sollievo della Sofferenza,’ Viale Padre Pio, 71013 San Giovanni Rotondo (FG)
Italy
Source of Support: None, Conflict of Interest: None
DOI: 10.4103/1477-3163.78279
Abstract
Colorectal cancer (CRC) is a major health problem in industrialized countries. Although inflammation-linked carcinogenesis is a well accepted concept and is often observed within the gastrointestinal tract, the underlying mechanisms remain to be elucidated. Inflammation can indeed provide initiating and promoting stimuli and mediators, generating a tumour-prone microenvironment. Many murine models of sporadic and inflammation-related colon carcinogenesis have been developed in the last decade, including chemically induced CRC models, genetically engineered mouse models, and xenoplants. Among the chemically induced CRC models, the combination of a single hit of azoxymethane (AOM) with 1 week exposure to the inflammatory agent dextran sodium sulphate (DSS) in rodents has proven to dramatically shorten the latency time for induction of CRC and to rapidly recapitulate the aberrant crypt foci-adenoma-carcinoma sequence that occurs in human CRC. Because of its high reproducibility and potency, as well as the simple and affordable mode of application, the AOM/DSS has become an outstanding model for studying colon carcinogenesis and a powerful platform for chemopreventive intervention studies. In this article we highlight the histopathological and molecular features and describe the principal genetic and epigenetic alterations and inflammatory pathways involved in carcinogenesis in AOM/DSS-treated mice; we also present a general overview of recent experimental applications and preclinical testing of novel therapeutics in the AOM/DSS model.
Keywords: Animal model, chemical carcinogens, colorectal carcinogenesis, preclinical studies
How to cite this article:
Robertis MD, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J Carcinog 2011;10:9
How to cite this URL:
Robertis MD, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J Carcinog [serial online] 2011 [cited 2021 Oct 15];10:9. Available from: https://carcinogenesis.com/text.asp?2011/10/1/9/78279
Introduction
Colorectal cancer (CRC) is the third most common malignancy in the world. This malignancy can develop spontaneously or as a late complication of a chronic inflammatory state. Many environmental causes of cancer and other risk factors are associated with some form of chronic inflammation. Up to 20% of cancers are linked to chronic infections, 30% can be attributed to tobacco smoking and inhaled pollutants (such as silica and asbestos), and 35% can be attributed to dietary factors (for example, 20% of the cancer burden is linked to obesity).
In the context of the chronic inflammation that precedes tumor development there is deregulation of the immune system. Inflammatory bowel disease (IBD) represents an example of a condition that greatly increases the risk of colorectal cancer. [1] Furthermore, the development of CRC is influenced by a genetic predisposition, which is especially high for somatic mutations of the tumor suppressor gene adenomatosis polyposis coli (APC) that causes familial adenomatosis coli syndrome. [2]
A variety of available CRC animal models have provided important tools for investigating the complex development and pathogenesis of CRC. These models can be used to provide new insights into the etiologic and pathophysiologic mechanisms as well as the treatment of human CRC. For example, the animal models help us to understand the sequential acquirement of genetic and epigenetic alterations, with consequent changes in cell behavior and tumor biology, that is observed in man. A number of models which display the metastatic process and the characteristic sensitivity to therapeutics have also been provided. [3] Animal studies cannot replace human clinical trials, but they must be opportunely used in preclinical studies so that human diagnostic- and therapeutics-oriented trials can become more focussed, with greater chances of success. Indeed, these models are not only precious instruments for uncovering new mechanisms of CRC pathophysiology but are also promising tools for advancing our understanding of the tumor response to novel chemopreventive and therapeutic strategies.
There are numerous CRC animal models that approximate some of the characteristics of human CRC, each with its own peculiar advantages and limitations. Thus, any specific experimental issue should be studied by choosing the model best suited to resolve particular tasks. In this review, different rodent models developed in the last few years for the study of CRC are described, with an extensive overview of the currently most commonly used model of chemically induced colon carcinogenesis, the AOM/DSS model. In this article we compare the different protocols of tumor induction, present a comprehensive description of the histopatologic and molecular features of the AOM/DSS model and discuss its main advantages and limitations, as well as its recent experimental applications.
Experimental Colon Carcinogenesis in Rodents
CRC development is a long-term process, from normal epithelial cells via aberrant crypts and progressive adenoma stages to carcinomas in situ and then metastasis. A variety of models of CRC have been developed that mimic the etiology and pathobiology of human tumors; both in vitro and in vivo models are available to reflect every stage of CRC development.
In vitro models permit isolation of specific aspects of tumor biology, such that functional analysis of relevant genes or the assessment of effects of endogenous mediators and pharmacological compounds are faster and simpler than in the intact organism. However, in vitro models can reflect only specific stages of tumor development, depending on the stage they have originated from. For cell lines, this limitation restricts the model to tumor stages with sufficient intrinsic growth potential, i.e., carcinoma and, with much more difficulty, adenoma. In contrast, primary cultures that still largely resemble the cell population in vivo can be obtained from any stage, including normal epithelium. [4] On the other hand, the major disadvantage of in vitro models when investigating a complex disease like CRC is the loss of tissue context that affects tumor growth and metastasis in vivo. By contrast, the ability to reliably induce colon tumors in in vivo models provides us the opportunity to study various aspects of the carcinogenesis or metastatic process as seen in human CRC but, usually, not both. Many rodent models have been developed to evaluate various features of CRC in humans. Some of these models date back almost 80 years. These models have provided information on the initiation, promotion, and progression of tumors. Oncogenesis studies using these models have elucidated the role of genetic and environmental factors and of other influences on the various aspects of this complex disease. The established models can be used for chemoprevention studies as well as to evaluate immunological, chemical, and surgical therapy regimens. Rodent models of CRC may be a) genetically modified animals (e.g., Min/ΔAPC-mouse strains, which reproduce tumor development, starting from initiated (mutated) epithelial cells to cover polyp growth through tumor progression and, rarely, metastases; b) xenoplant models, which reflect tumor growth and metastasis (only feasible using malignant cells that have sufficient intrinsic growth potential to form tumors in animal hosts); or c) chemically-induced models (chemical induction of tumors acts on the normal epithelium to form carcinomas via the multistage process previously described and only rarely metastases).
Genetic models
Mutant mice carrying a heterozygous germline mutation in the APC gene (Min/ΔAPC-mouse), similar to that found in familial adenomatous polyposis (FAP) patients, have been obtained. [5] These animals develop multiple intestinal neoplasia (Min-mouse); they mimic the rapid tumor development seen in FAP patients and model the process of polyp growth and progression. In the tumors, the loss of the APC allele, which normally prevents binding and degradation of β-catenin, constitutively activates the Wnt-pathway and deregulates growth in tumor cells. K-ras mutations and inactivation of p53 have not been observed in Min-mouse tumors. [5] Different ΔAPC-mouse strains have been used for functional analysis of the APC gene product and mapping of essential domains. [5] In addition, double mutant mouse strains have been constructed that carry both a mutated APC gene and alterations in modifier genes. For example, mice with homozygous deletions of phospholipase A2, [6] cyclooxygenase-2 (COX-2), [7] and prostaglandin-receptors EP2 and EP4 [8] present impaired neovascularisation of polyps and attenuated tumor development, which has also been observed in human colorectal polyps. Inactivation of Smad3, a frequent defect in human CRC, causes accelerated tumorigenesis and metastasis to the lymph nodes. [5],[9]
The one striking difference between Min/ΔAPC-mice and human FAP patients is tumor location, which is predominately in the small intestine and extremely rare in the colon of Min-mice [10] whereas, by contrast, FAP patients while also developing small intestinal tumors, carry large numbers of adenomatous polyps in their colon and rectum.[11] In spite of this, Min-mouse models are widely used in the search for nutritional risk factors and chemopreventive compounds, usually by adding the test compound to the animal’s diet.
Construction of mouse models for hereditary nonpolyposis colorectal cancer (HNPCC) has been less successful as the heterozygous deletion of the mismatch repair genes involved (Msh2, Mlh1, Msh6, Msh3, Pms2, and Pms1) did not prove to be sufficient to predispose to cancer. Consequently, mice carrying homozygous deletions have been used as disease models for HNPCC-like cancers. [9] Although such mice are more susceptible to tumor formation, the tumor spectrum observed consists of various lymphomas that are almost never encountered in HNPCC affected patients. Gastrointestinal tumors are only observed in animals that do not succumb to lymphomas in early life. Deletions in Msh2 and Pms2 potentiated APC-mediated intestinal tumorigenesis. [9]
Xenoplant models
The ability of tumor cells to develop tumors after subcutaneous or intravenous injection in immunodeficient mouse strains (nude, bg/nu/xid, or SCID mice), allowed the analysis of human tumors in vivo.[12] As the xenoplant model is straightforward and easily achievable, it has been used to supply human tumors in an experimental in vivo environment for the assessment of cytostatic therapeutic compounds. Although the introduction of xenoplant models represents an important step towards a pathobiologically relevant model of tumor metastasis, subcutaneous or intravenous injections of tumor cells establish a model that neglects the complexity of tumor growth and metastasis and the interactions between tumor and microenvironment, which largely controls the biological characteristics of tumors. Orthotopic transplantation into the cecum and rectum has been developed to overcome this problem and create a clinically accurate model for human CRC. From orthotopically-transplanted tumors, metastasis occurs to local lymph nodes and liver as expected. [13] However, an important practical limit of orthotopical transplant is the complexity of the surgical procedure involved, along with the lack of immune function that is a necessary element of all xenoplant models. These limitations eliminate the principal mechanisms that govern the effects of inflammation and immunity on tumor development.
Chemically induced models
Carcinogen-induced colon cancer in rodents can recapitulate in a highly reliable way the phases of initiation and progression of tumor that occur in humans. Such models are frequently used to assess activity of chemopreventive compounds and to identify risk factors.
Even if the high carcinogen dose given in experimental settings differs from the common situation in humans, where tumors arise from exposure to small doses of carcinogen present in the typical Western-style diet plus additional environmental factors, there are a number of advantages in studying the pathogenesis of carcinogen-induced colon cancer in rodents. These models are highly reproducible, they can be readily tested on animals with different genetic backgrounds, and the pathogenesis can recapitulate human CRC.
A variety of chemical agents have been used to induce colon tumors in animals. They include direct- and indirect-acting agents, according to the requirement of biological catalysis to form the ultimate reactive species. Unlike direct-acting agents, the indirect-acting carcinogens require enzymatic action to be converted into the active species which are able to alter cellular macromolecules such as nucleic acid or proteins.
Chemical carcinogens widely used to induce colon lesions similar to human malignancy are: (a) heterocyclic amines, (b) aromatic amines, (c) alkylnitrosamide compounds, and (d) dimethylhydrazine and azoxymethane. The carcinogenic properties of these substances and their effects on histogenesis, cell proliferation kinetics, interaction between genetic susceptibility traits, and enviromental factors have been reported.
Heterocyclic aromatic amines: After the identification of heterocyclic aromatic amines in grilled or broiled meat, the main representatives of this group of carcinogens, i.e., 2-amino-33-methylimidazo [4,5-f]quinoline (IQ) and 2-amino-1-methyl-6-phenylimidazol [4,5-b]pyridine (PhIP), have been introduced into experimental CRC.
These compounds show a multi-target-organ specificity, with induction of lesions also in the mammary gland and prostate of rodents. [14] Liver metabolic activation of IQ to its ultimate carcinogen is required prior IQ-DNA adduct formation which has been reported in different organs. Metabolic activation of PhIP proceeds through steps involving enzymes of phase I and phase II metabolic reactions, mainly in the liver and in the target organs. Studies on PhIP activation and detoxification suggest that repeated exposures in colonic mucosa are required to activate PhIP in vivo.[15] Dietary administration of IQ and PhIP in rodents for 52 weeks resulted in a low tumor incidence (5%-28%), [16] whereas with a PhIP-feeding regimen for 104 weeks [14] colon tumor incidence (43%-55%) was associated with severe toxicity.
Moreover, combining PhIP with a high-fat diet or with AOM [17] causes enhancement of mutagenic and tumorigenic effects. PhIP induces a higher frequency of mutations in APC as well as microsatellite instability, but causes neither K-ras nor p53 mutations. [18]
Aromatic amines: Chemical induction of both benign and malignant epithelial neoplasms in rat and in rodent models with 3,2′-dimethyl-4-aminobiphenyl (DMBA) has been described. [19] However, DMBA tumorigenic activity is less potent than that with the series of compounds derived from the other chemical carcinogens. Two important disadvantages of this compound are: (i) multiple injections of DMAB are required to induce colon tumors and (ii) there is induction of neoplasms in other tissues. [20]
Alkylnitrosamide compounds: Methylnitrosourea (MNU) and N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) are alkylating carcinogens that require an intrarectal injection to induce lesions in the distal colon and rectum in rodent models. [21] Because biochemical activation is not required, these compounds might be ideal topical carcinogens. On the other hand, the mode of administration represents their weakness.
Dimethylhydrazine and metabolic derivatives: Classically, 1,2-dimethylhydrazine (DMH) or its metabolite azoxymethane (AOM) have been used to induce CRC in mice and rats. DMH is a precursor of methylazoxymethanol (MAM), a carcinogen found in cycad flour. [22] Both DMH and AOM require several metabolic activation steps (including N-oxidation and hydroxylation) to induce DNA-reactive adducts. Hydroxylation of AOM results in the formation of the reactive metabolite MAM, which can alkylate macromolecules in the liver and colon, and operate the addition of methyl groups at the O6 or N7 position of guanine (O6-methyl-deoxyguanosine and N7-methyl-deoxyguanosine) in the DNA molecule. Methylation at the O6-position of guanine has been shown to be the primary promutagenic lesion produced by AOM. [23] Other enzymes are involved in the metabolic activation of AOM. An in vitro study suggested that MAM activation in mouse is independent of colon alcohol dehyidrogenase. [24] On the other hand, the alcohol-inducible cytochrome P-450 isoform, CYP2E1, plays an important role in the conversion of AOM to MAM. [25]
In the earliest published studies repetitive administration of DMH in rodents was demonstrated to induce colon tumors with pathological features similar to that seen in sporadic human disease, [26],[27] Later studies switched to AOM, [28],[29],[30] due to practical advantages such as reproducibility, high potency, simple mode of application, excellent stability in solution, and low price. [29]
DMH- or AOM-induced tumors share many of the histopathological characteristics of human CRC and they frequently carry mutations in K-Ras and β-catenin; some show microsatellite instability indicative of a defective mismatch repair (MMR) system. APC mutations, however, are less frequent in rodents; p53 mutations are rarely observed; and the tendency to metastasize is low as has been summarized by Kobaek-Larsen et al. [18]
Comparison of Different AOM/DSS Protocols in Different Rodent Strains
The combination of AOM with the inflammatory agent DSS for induction of CRC in rodents has proven to dramatically shorten the latency time observed in the classical model of spontaneous CRC, which is based on several repeated intraperitoneal (i.p.) AOM injections. The administration of AOM with DSS in drinking water causes rapid growth of multiple colon tumors per mouse within 10 weeks, whereas there is a latency time of 30 weeks in the other models. [29] The AOM/DSS model is also particularly applicable when the focus of the study is on tumor progression driven by colitis, as in ulcerative colitis (UC) or Crohn disease (CD).
Experimental CRCs develop in the colon of AOM/DSS-treated mice from aberrant crypt foci (ACF) [31] and adenomas, which are both induced in a dose-dependent manner. Depending on the mouse/rat strain, the dose, and the regimen of application, the latency time is approximately 20 and 50 weeks, respectively, for mouse and rat models [28],[32] [Figure 1].
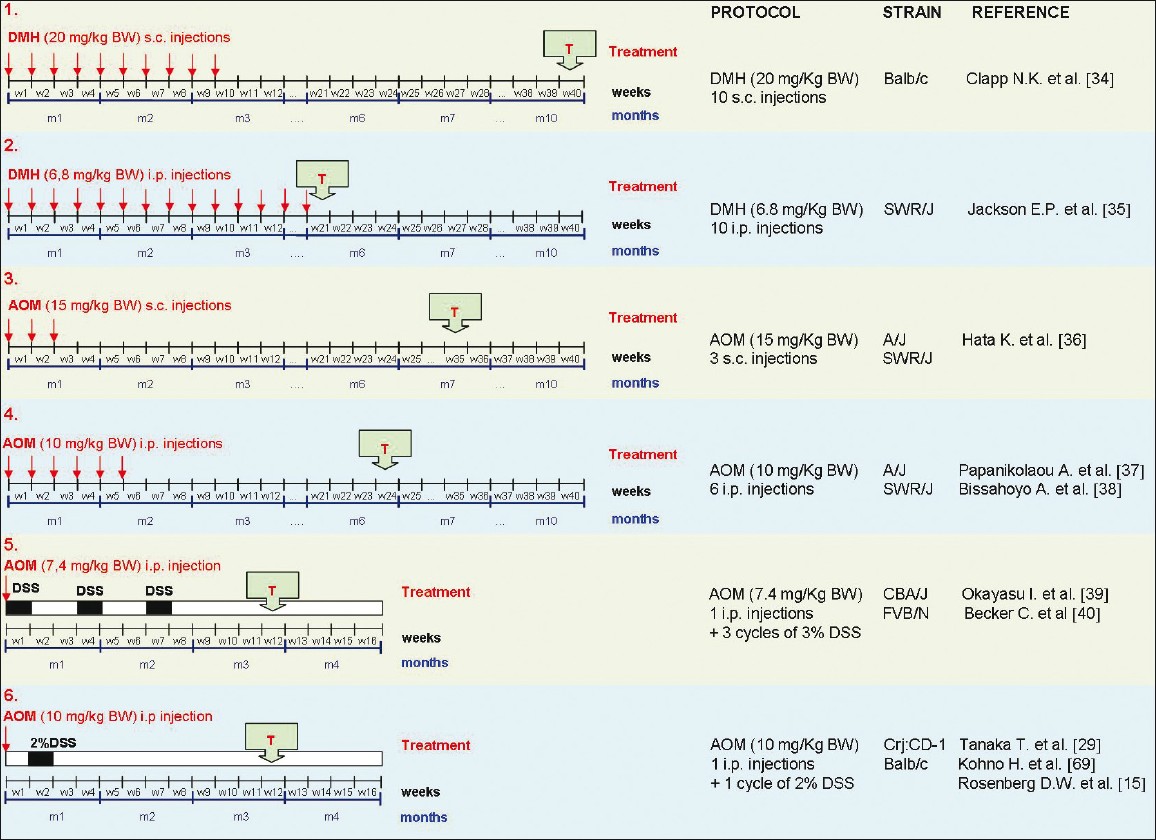 |
Figure 1: Comparison of different protocols based on DMH-AOM-DSS administration for induction of colorectal tumors in the murine model. Different protocols based on DMH or AOM, administered individually or in combination with DSS, are schematically represented. The time of tumor identification (T) has been indicated. Protocol number 6 shows a short latency time and a very simple procedure of tumor induction. Click here to view |
Genetic susceptibility of rodent strains
As in the human population, the genetic background of rodent strains has been demonstrated to play an important role in the risk for colorectal tumorigenesis. Notably, genetic heterogeneity has been found to be a relevant modifier of AOM-induced tumorigenesis, which is reflected by the wide differences in susceptibility to AOM between different mouse and rat strains.
Several studies to investigate the susceptibilities of different murine strains to AOM/DSS treatment have been conducted. Suzuki et al. [30] demonstrated differential sensitivities to the AOM/DSS protocol among four different murine strains (BALB/c, C3H/HeN, C57BL/6N, and DBA/2N). The protocol used in this study included a single i.p. injection of AOM (10 mg/kg body weight), followed by 1% (weight/volume) DSS in drinking water for 4 days. There was 100% tumor frequency in Balb/c mice and only 50% in C57BL/6N mice. C3H/HeN and DBA/2N mice did not develop adenocarcinomas but only a few adenomas. Interestingly, in mice subjected to AOM injection followed by 0.25% or 0.1% of DSS in drinking water, only dysplastic crypts were observed and no colon tumors were reported.
Several studies have also been done to clarify the differences in susceptibility of rats with different genetic backgrounds. Morten Kobaek-Larsen et al. [18] reported a strain-dependent susceptibility to AOM among rat strains: AOM-treated BDIX/OrlIco rats showed a higher incidence of tumor formation (75%-100%), with high reproducibility and a relatively short latency time (6-8 months), compared to two other inbred rat strains (F344/NHsd and WAG/Rij). In this study, BDIX/OrlIco rats (9-12 weeks old) were subjected to four subcutaneous (s.c.) weekly injections of AOM (15 mg/kg body weight) with a week off after the first two treatments.
Historically, the majority of colon carcinogenesis studies have been carried out in rats. However, the possibility of reliably generating tumors with high frequency within the distal colon of mice, as well as the histogenesis of multiple adenomas with subsequent development of adenocarcinomas in carcinogen-treated colon of mice, endorses the importance of the murine species for studying the pathogenesis of colon cancer. Recently, the scientific community has largely switched to mouse models of colon carcinogenesis. This is due to the advantages of murine models, for example, the availability of extensive genetic information on individual mouse strains, the existence of recombinant inbred mouse panels, and the ever-increasing number of transgenic, knockout and knockin genetic models that are available for study.
Comparison of the AOM/DSS protocols in mice
Numerous protocols with AOM and DSS, in combination or alone, have been used in different mice strains. Here we report the most commonly used protocols of tumor induction, describing treatments based on repetitive administrations of only DMH or AOM, as well as colitis-associated CRC models based on the AOM/DSS protocol. As shown in [Figure 1], when AOM is combined with the proinflammatory stimulus DSS, tumor growth is accelerated and there is a significantly shorter latency time (2-3 months).
BALB/c mice treated with 10 s.c. injections of DMH (20 mg/kg body weight/week) and sacrificed 40 weeks after the first DMH injection have been shown to develop tumors with high incidence, almost exclusively in the distal colon. [33]
SWR/J mice treated with increasing i.p. doses of DMH (6.8, 34, 68, and 136 mg/kg body weight) showed a dose-dependent decrease in survival time. [35] Four different protocols based on i.p. injections of DMH (6.8 mg/kg body weight/week) were tested, giving the carcinogen once a week for 1, 5, 10, or 20 weeks. At 20 weeks, colon tumors developed in 26%, 76%, and 87% of mice given a total dose of 34, 68, and 136 mg/kg DMH, respectively; no tumors were detected in animals treated with a total dose of 6.8 mg/kg. Most colon tumors (79%) were located in the distal colon, with the remainder being found in the mid-colon; no tumors were detected in either the proximal colon or the small intestine.
SWR/J mice subjected to 3 s.c. injections of AOM (15 mg/kg body weight/week) and sacrificed at 35 weeks after the start of treatment have been reported to show a high incidence of colonic tumors. [35] Interestingly, although AKR/J mice had a lower incidence of colonic tumors at 35 weeks than SWR/J mice, the frequency of ACF was similar in AOM-treated AKR/J and AOM-treated SWR/J mice. In both strains, ACF were detected at high frequency in the proximal colon, whereas tumors arose mainly in the distal colon.
A/J or SWR/J mice treated with 6 repeated i.p. injections of AOM (10 mg/kg body weight/week) and killed at 1, 2, 4, 6, 9, and 24 weeks after the last injection showed high incidence of colorectal tumors at week 24. Neoplasm formation could be revealed as early as 4 weeks after AOM exposure. [36]
Bissahoyo et al. suggested that 4 injections might be sufficient for a high tumor induction rate (~90%) in the highly susceptible A/J strain but not in moderately susceptible ones such as the SWR/J strain. [37]
Female CBA/J subjected to 1 i.p. injection of AOM (7.4 mg/kg body weight) + three exposition cycles to DSS (each cycle included administration of 3% DSS in drinking water for 7 days, followed by distilled water for 14 days) showed a latency time of 12 weeks. [38] Multiple mucosal tumors (10.5/mouse) in combination with chronic colitis developed in the colorectum. More recently, similar results have been reported in FVB/N mice. [39],[40] This model reproduces a state of chronic inflammation, which is believed to be the driving force in tumor progression in colitis-associated CRC (e.g., as in UC or CD). [29],[38],[39]
Crj:CD-1 (ICR) mice (outbred strain) [28] receiving 1 i.p. injection of AOM (10 mg/kg body weight/week) followed by DSS (2%) in drinking water for 7 days (1 week after the AOM injection) showed 100% incidence of adenocarcinomas at 20 weeks after the first treatment, whereas another study revealed the presence of tumors already at week 6. [15] Four other inbred murine strains (Balb/c, C57BL/6N, C3H/HeN, and DBA/2N) were used in the same experimental conditions. Among these, Balb/c mice displayed the same findings as was obtained with the ICR strain, whereas C57BL/6N, C3H/HeN, and DBA/2N were demonstrated to be more resistant to AOM.
AOM/DSS Model of Colorectal Carcinogenesis
A number of experiments have demonstrated the relevance of the AOM/DSS mouse model in the study of the mechanisms of human colorectal carcinogenesis. Due to the synergic effects of AOM (tumor-inducing agent) and DSS (tumor-promoting agent), the AOM/DSS model reproduces colorectal carcinogenesis promoted by an initial acute inflammation phase, showing a shorter latency period than models based on only AOM or DSS administration. For instance, oral administration of a DSS solution to rodents, a widely used IBD mode, typically requires a long time exposure or many cycles of DSS administration and, moreover, the incidence of tumors is relatively low. [41]
As described by Tanaka et al. [28] the AOM/DSS model is based on a single i.p. injection of AOM (10 mg/kg body weight) and a single cycle of the inflammatory agent DSS (2%) in drinking water. With this protocol there is rapid development of multiple colon tumors in the outbred strain (crj:CD1) as well as in the inbred strain (Balb/c) within approximately 12 weeks. DSS dissolved in drinking water is toxic to the epithelial lining of the colon and produces severe colitis, which is characterized by loss of body weight and bloody diarrhea in the first weeks following DSS consumption; however, these phenomena decrease significantly after 5 weeks from the start of AOM/DSS treatment.
Development of cancer in this model closely mirrors the pattern seen in humans. Tumors induced in mice exposed to AOM/DSS treatment accurately recapitulate the pathogenesis observed in human CRC. For example, tumors are very frequent in the distal part of the colon, which is also the predominant location of spontaneous CRC in man. They often start with a polypoid growth and frequently exhibit histopathological features similar to human CRC. However, AOM-induced tumors often lack mucosal invasiveness. [42] Although AOM-induced colon tumors in mice and rats have a very low tendency to metastasize, the analysis of lymph nodes and other peripheral organs by macroscopic and microscopic techniques might be considered for studies examining factors potentially involved in metastasis.
Histogenesis of CRC in the AOM/DSS model
Colon cancer is thought to develop by a multistep process in which normal crypts are initiated to form foci of aberrant crypts (ACF) that proliferate by crypt fission to form microadenoma. The microadenomas enlarge to give macroscopic adenomas, adenomatous polyps, and finally adenocarcinomas. Thus there is morphological and genetic progression through an adenoma-carcinoma sequence. [43]
Such multistep tumor development can be efficiently reproduced in the AOM/DSS murine model with high reproducibility in both inbred (Balb/c) and outbred [crj:CD-1 (ICR)] susceptible murine strains [Figure 2]. Usually, 3-10 macroscopic tumors develop in 80%-100% of the animals, with tubular adenoma, dysplasia, and colitis with mucosal ulceration being present in male mice of susceptible strains at week 12 from the start of the treatment. [37],[40]
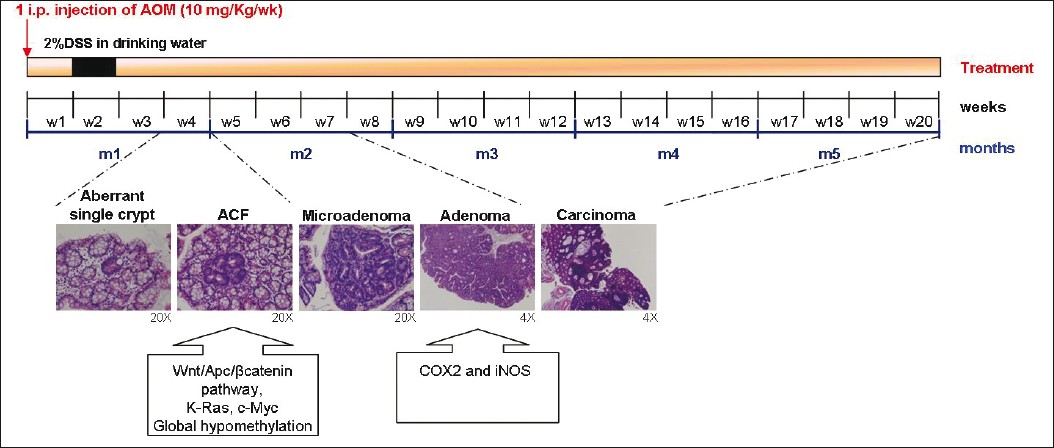 |
Figure 2: Schematic representation of multistep tumor progression in the AOM/DSS murine model. The multistep tumor progression observed in the AOM/DSS murine model of CRC, based on the ACF-adenoma-carcinoma sequence, is represented on the timeline, with the principal molecular alterations assessed in specific phases of the carcinogenic process. Click here to view |
Colonic crypts can easily be observed by examining the mucosal surface of the colon after it has been opened and fixed and stained with methylene blue. When examined in this way, colons of animals that have been treated with AOM/DSS 3-4 weeks earlier are found to have occasional, abnormally large, darkly staining, and slightly raised ‘aberrant crypts’ [Figure 3]. With longer time intervals between carcinogen administration and observation, these aberrant crypts are frequently found as foci, with two to many hundreds of aberrant crypts appearing together in a cluster. Many aberrant crypts on histological examination show a high level of dysplasia and are then correctly called microadenoma (more than 10 crypts, each less than 1 mm in size). [44] As microadenomas grow, they might evolve to macroscopic adenoma and, eventually, colon cancer [Figure 3].
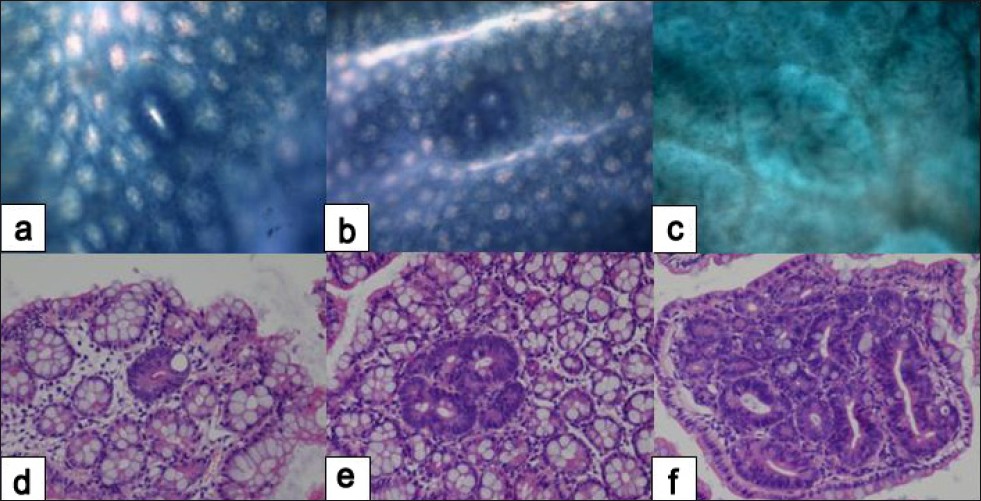 |
Figure 3: Histopathology of early colonic neoplasms developed in AOM/DSS-treated mice. Single aberrant crypt (a, d); aberrant crypt focus – ACF (b, e); microadenoma (c, f). Methylene blue (a, b, c) and hematoxylin-eosin (d, e, f) stain. Original magnification, (a, b, c) ×10; (d, e, f) ×20. (Unpublished data). Click here to view |
AOM/DSS-induced tumors typically display adenomatous growth before showing malignant degeneration and invading the bowel wall, and they are frequently infiltrated by T lymphocytes and other immune cells. On macroscopic examination, nodular, polypoid, or caterpillar-like colonic tumors are exclusively observed in the middle and distal colon of AOM/DSS-treated mice. According to Ward, histologically these tumors can be tubular adenoma or well/moderately-differentiated tubular adenocarcinoma. [45] As demonstrated by Tanaka et al., [28] the incidences of adenocarcinoma and tubular adenoma at the 20 th week is 100% and 38% with a multiplicity of 5.60±2.42 and 0.20±0.40, respectively. Similarly, there is an incidence of 100% of adenomas and 50% of adenocarcinomas as early as week 8, with a multiplicity of 4±1.6 and 0.3±0.7, respectively (De Robertis, unpublished data). After 21 weeks from the start of treatment, only few adenocarcinomas are observed to invade the submucosa, muscularis propria, or serosa (De Robertis, unpublished data) [Figure 4].
 |
Figure 4: Macroscopic observation and histopathology showing late colonic neoplasms developed in AOM/DSS-treated mice. Tubular adenoma (a, d); moderately differentiated adenocarcinoma (b, e); and moderately differentiated adenocarcinoma invading into the mucosa (c, f). Macroscopic analysis in necroscopy (a, b, c) and hematoxylin-eosin stain (d, e, f). Original magnification ×4. (Unpublished data). Click here to view |
Microscopically, these tumors display the characteristics of intraepithelial neoplasia, with changes ranging from low-grade dysplasia (usually located at the periphery of polyps, next to non-dysplastic colonic epithelium) to high-grade dysplasia/intramucosal carcinoma (predominantly forming the central parts of the tumors and infiltrating to the submucosa).
Colonic mucosal dysplasia (low- and high-grade) has been described by Riddell [46] and Pascal. [47] Specifically, hyperplasia (with no dysplasia) shows slightly dilated crypts with a normal epithelium. In mild dysplasia the nuclei are elongated, slightly crowded, and pseudostratified, but their polarity is well preserved and the number of goblet cells is normal or slightly reduced. In moderate dysplasia the nuclei are elongated and more crowded and also more pseudostratified than in mild dysplasia, but their polarity is still well preserved; the number of goblet cells is more reduced than in mild dysplasia. In severe dysplasia the nuclei are enlarged, round or ovoid, with prominent nucleoli; also, nuclear polarity is partly lost, numerous mitoses are present, and the number of goblet cells is markedly reduced or the cells are completely lost.
Colonic mucosal ulceration with focal dysplasia can be found in the distal colon of mice. Colitis is usually graded on hematoxylin-eosin stained sections as showing a) normal appearance (grade 0); b) mild infiltration of lamina propria by inflammatory cells, with no erosion (grade 1); and c) deep erosion with marked inflammation, often including crypt abscess formation (grade 2). [28] Mucosal ulceration (grade 2) and colitis with mild erosion can be found in mice at 2 weeks from the end of DSS administration, but the inflammatory state progressively decreases in the following weeks and a resolution of the acute inflammation can be observed at week 6 (De Robertis, unpublished data).
A particular focus on precancerous lesions observed in the AOM/DSS model
It is generally accepted that colon carcinogenesis, irrespective of animal species, is a focal and sequential process that originates from a single colonic crypt. The growth and the morphological and molecular features of ACF support the contention that ACF are preneoplastic lesions and so the traditional ‘adenoma-carcinoma’ sequence of colorectal carcinogenesis has been extended to the ‘ACF-adenoma-carcinoma’ sequence. [48]
In humans, ACF were described and partially characterized for the first time in 1991. [49] In 1994, Pretlow first detected altered enzymatic activity and crypt dynamics and proliferation. [50] ACF can be observed in patients with both sporadic CRC and FAP. [51] They closely resemble the aberrant crypts seen in rodents treated with carcinogens.
Identification of ACF both in human colon and in AOM/DSS-treated rodents makes the study of CRC at the precancerous stages possible with this model. In 1987, Bird and Good [31] described ACF as preneoplastic lesions in the colon of carcinogen-treated rodents. Because classical ACF protrude toward the lumen they can be easily scored by light microscopic surface examination of formalin-fixed whole-mount colon preparations stained with methylene blue.
Since the total number and size (crypt multiplicity) of these small lesions can be scored easily, ACF have been used as a short-term bioassay to evaluate the role of nutritional components and chemopreventive agents at an early stage of colon carcinogenesis. Under the microscope the aberrant crypts are larger and have a thicker epithelial lining than normal crypts, and are usually gathered together in foci of one to many hundreds of aberrant crypts. These aberrant crypts sometimes are slightly elevated above the surrounding normal mucosa and often have slit-like lumens in crossection. [44],[49]
At weeks 3-4 from the start of the AOM/DSS protocol, surface examination of the luminal mucosa of colon preparations stained with methylene blue reveals flat ACF, classical elevated ACF, and nascent tumors [Figure 3]. Since flat ACF are not observed as elevated structures, their bright blue appearance and the compressed pit pattern of the crypt openings seen with transillumination could be used as criteria for their identification. Flat ACF and nascent tumors display a uniform picture of severe dysplasia, compressed pit pattern, overexpression of cytoplasmic/nuclear β-catenin, and nuclear overexpression of cyclin D1. [52] Apparently, flat ACF and tumors represent the same type of dysplastic lesions at different stages of crypt multiplication. In contrast, classical elevated ACF do not seem to be as clearly related to tumorigenesis. Furthermore, flat ACF have been shown to grow significantly faster than classical elevated ACF. [52]
Although ACF share many morphological, genetic, and biochemical features with colonic tumors, the association between ACF formation and tumorigenesis is not a simple one. Importantly, in carcinogen-treated rodents, a negative correlation between ACF formation and tumor formation and a discrepancy between distribution of ACF and distribution of tumors [53] has been reported. In AOM- or DMH-treated rodents and in patients with sporadic CRC, the number of tumors is minuscule in comparison with the large number of ACF, indicating that only a very small fraction of ACF has the potential to progress to the stage of a tumor. [51] This is consistent with the observation that a large fraction of ACF is hyperplastic, whereas only a small fraction of ACF shows dysplasia, the hallmark of malignant potential. Recent findings of the presence of β-catenin accumulated aberrant crypts (BCAC), [54] mucin-depleted crypts (MDF), [55] and flat-dysplastic ACF (FDACF) [56] in AOM/DSS-treated rodents are significant advances in the identification of events leading to tumor development. As described earlier, the morphological features of these lesions resemble those of advanced ACF, strongly suggesting that they may be subtypes of advanced ACF. However, the exact evolutionary sequence of BCAC, MDF, or FDACF and their relevance to the human situation is still unclear. These specific ACF-subtypes are purported to accurately predict tumor outcome. [51] Indeed, the simple methylene blue staining method for assessing ACF in whole mounts of colon remains a valuable tool when screening compounds for their colon cancer chemopreventive potential. However, their use as biomarkers in the screening of chemicals/agents for potential colon cancer chemoprevention action requires further scrutiny.
Molecular Features of The AOM/DSS Model and Relevance to Human CRC
Genetic pathways involved in the AOM/DSS model
Studies in mice and rats have revealed that AOM/DSS-induced tumors display very similar features to human CRC even at the molecular level. Such tumors display evidence of dysregulation of the APC/β-catenin-signaling pathway, such as aberrant protein expression of APC plus mutations and altered cellular localization of β-catenin. [57],[58],[59] In addition, the target genes of the APC/β-catenin signaling pathway, represented by cellular myelocytomatosis oncogene (c-Myc), cyclin D1, and cyclin-dependent kinase 4 (Cdk4), have also been shown to be dysregulated. [59] Consistent with findings in human CRC, AOM/DSS-induced tumors also have mutations of K-Ras and increased levels of enzymes involved in prostaglandin and nitric oxide synthesis, such as cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS). [58],[60]
In a recent study, a comprehensive protein expression analysis in the AOM/DSS-induced mouse colon carcinogenesis model demonstrated that a number of proteins functionally related to metabolism, antioxidant system, oxidative stress, mucin production, and inflammation are differentially expressed in the cancerous tissues of mice receiving AOM and DSS. [61] The number of proteins with altered expression was much smaller than the number of genes that showed differential expression patterns in a previous DNA microarray analysis. [62]
Wnt/APC/β-catenin pathway
The Wnt signaling pathway, which is normally involved in repressing differentiation during embryogenesis and in the postembryonic regulation of cell positioning in the intestinal crypts, is known to be involved in tumor formation when aberrantly activated. [63] β-Catenin is an important molecule in the Wnt-APC signal transduction system and a key component of the cadherin-mediated cell-cell adhesion system. It binds to the T-cell factor-4 (Tcf-4) in the nucleus and thereby regulates transcription of genes related to the growth, development, and differentiation of colonic cryptal cells. Accumulation of cytoplasmic and nuclear β-catenin, which may result from mutations in either APC, β-catenin, or Axin genes, or in general from activation of the Wnt signaling pathway, is associated with colon carcinogenesis.
As in human CRC, mutation of β-catenin is reported to be an early event of murine colorectal carcinogenesis. Inactivating APC/Apc mutations or activating β-catenin mutations, which mimics Wnt stimulation and leads to β-catenin accumulation, is observed in the majority of colon cancers in both humans and rodents. [64],[65]
Tumors in the AOM/DSS carcinogen model, similarly to the Apc Min/+ model, have signaling alterations marked by the activated canonical Wnt pathway. [37] Immunohistochemically aberrant expression of β-catenin has been reported in colonic neoplasms and dysplasia in inflammation-related colonic cancer in rodents [65] and humans. [66] The presence of β-catenin gene mutations might explain the altered immunohistochemical stainability of β-catenin in colonic neoplasms as well as dysplasias developed within 12 weeks after the start of the AOM/DSS treatment. [28] In contrast to human colon cancers without APC mutations, where the β-catenin gene is frequently mutated at codons 33, 41, and 45 of the glycogen synthase kinase-3β (GSK-3β) phosphorylation motif, mutations in AOM/DSS-induced colon adenocarcinomas are found at codons 32, 33, and 34. [67] This location is slightly different from that mentioned in another report, where β-catenin gene mutations of murine tumors induced by AOM were present at codons 33, 34, 37, and 41, but not at codon 32. [65] In addition, β-catenin gene mutations have been detected at codon 32, 34, 37, and 41 in DMH/DSS-induced colon adenocarcinoma. [68] Koesters et al. reported that the β-catenin mutations at codons 37 and 41, which are important serine and threonine sites for GSK-3β phosphorylation, possess higher oncogenic potential.[69] They also demonstrated that the different mutational spectra observed in the β-catenin gene directly relates to the particular carcinogenic treatment given. Therefore, it has been speculated that DSS exposure causes a shift in the mutation sites from that induced by treatment with DMH or AOM alone.
Finally, a large upregulation of the Tcf-4 transcript can be also observed in AOM/DSS-induced tumors. [70]
K-Ras
Activating mutations in the K-Ras gene have been identified in a great variety of human carcinogenesis. The mutated forms are found to stimulate cell proliferation, transformation, and differentiation. [71] As in human CRC, K-Ras mutations are involved in the early stages of AOM/DSS colorectal carcinogenesis and are associated with increased tumor size. [72]
Consistent with findings in human CRC, K-Ras activation appears to play an important role both in the AOM and the DMH models, whereas this pathway is not frequently targeted by PhIP. Jacoby et al. [73] showed that DMH caused K-Ras-activating mutations (G to A) in 66% of colon mouse carcinomas.
p53 pathway
The growth-inhibitory p53 pathway is another important molecular checkpoint that is altered in the development of colitis-related colonic neoplasms of rodents and human. [74] In particular, the ARF/p53 pathway, which is effective at limiting the proliferation and survival of oncogenically transformed cells, appears to be dysregulated during the carcinogenic process in humans.
However, p53 immunohistochemical expression has not been detected in AOM/DSS-induced colonic dysplasia and neoplasms. [28] Similarly, no mutations of p53 have been found in colonic tumors developed in another experimental model of colitis-related carcinogenesis in rat. [75] This may be related to the absence or low frequency of p53 mutations in chemically-induced colon tumorigenesis in rodents.
Immunohistochemical observation revealed that only a few colonic tumors induced by DSS show positive reactivity against p53. [65] Recently, Nambiar et al. indicated that both ARF and p53 are sequence normal in AOM-induced colon tumors but, strikingly, the ability of p53 to activate or repress transcription is suppressed. [76] Importantly, it has been reported that nongenetic inhibition of p53 may play a critical role in the early stages of human colon carcinogenesis and in p53 sequence normal tumors. Thus, it is possible that the increased stability of p53 protein in the absence of sequence alterations may be a result of an aberrant acetylation and/or phosphorylation process. [77] Additional analyses are required to determine whether post-translational modifications are indeed involved in suppressing p53 activity in AOM-induced tumors.
c-Myc
Both Wnt/β-catenin and non-Wnt/β-catenin-mediated human CRCs display increased c-Myc expression. [79] c-Myc functions as a global mediator of the oncogenic process, linking together a seemingly heterogeneous pool of molecular mechanisms underlying cancer development. [78] The gene is a direct transcriptional target of the Wnt/β-catenin pathway, while the transforming growth factor-β (TGF-β) signaling is associated with c-Myc repression through SMAD3 binding to a repressive SMAD-binding element (RSBE) within the c-Myc promoter. [79] Numerous lines of evidence indicate that c-Myc may be a central mediator of CRC, perhaps through its role in chromatin remodeling. [78]
Mouse models have been used to show that decreased c-Myc expression leads to reduced numbers of CRCs, [80] a result confirmed by c-Myc inhibition in human CRC.[82] Recently, an increased expression of c-Myc and telomerase (TERT) have been observed in foci of high-grade dysplasia/intramucosal carcinoma isolated from AOM/DSS-treated Crj:CD-1 (ICR) mice. [62],[70] Of note, telomerase not only compensates for natural telomere erosion and thus contributes to permanent cell growth, but it is also able to activate progenitor cells by controlling c-Myc- and Wnt-related transcription pathways. [82]
TGF-β
TGF-β has long been implicated in the pathogenesis of sporadic CRCs, which often acquire resistance to TGF-β signaling. At later stages of cancer progression there is increased expression of TGF-β, promoting invasion and metastasis. [83]
Several TGF-β pathway-associated models have been used to dissect the complex role of this pathway during CRC development. TGF-β1-deficient mice die around 3 weeks of age due to extensive inflammation. [84] However, on a Rag2-deficient background, lacking functional B- and T-cells, TGF-β1-deficient mice survive until adulthood.[85] Mice deficient for both TGF-β1 and Rag2 rapidly develop carcinoma of the cecum and colon, suggesting that inflammation in combination with loss of TGF-β1 results in predisposition to cancer. [85]
While Tgfb1 is upregulated in the colonic mucosa of mice treated with AOM/DSS, Tgfb3 is a notable downregulated gene. [62] Decreased TGF-β3 mRNA level has been shown to be mediated by nitric oxide. [86] Becker et al. [87] investigated the functional role of TGF-β using the AOM model in TGF-β receptor-deficient and TGF-β transgenic mice, showing that the TGF-β receptor-deficient mice develop a significantly higher number of tumors than wild-type mice. Complementary data were obtained by Fantini et al. [88] who generated mice transgenic for the TGF-β inhibitor Smad7, with overexpression specifically on T-cells.
Cox-2
COX-2 is a cytoplasmic protein that catalyzes the synthesis of lipid inflammatory mediators (prostaglandins and prostacyclins) from arachidonic acid. Expression is increased at sites of inflammation as well as in ~80% of CRCs and 40% of colorectal adenomas. [89] The COX-2 protein is found in the cytoplasm of neoplastic colonic epithelial cells and to a lesser extent in stromal cells, whereas normal epithelium is negative for COX-2. After AOM/DSS challenge, the expression of COX-2 is upregulated in neoplastic cells in mice with mutations in the tumor suppressor Apc. [90]
COX-2 may contribute to tumor development by modulating apoptosis, angiogenesis, and tumor invasiveness. It also has a role in the progression of cancer via activation of metalloproteases, thereby increasing the invasiveness of colon cancer cells. [91]
The significant tumorigenic effect of COX-2 on the development of colorectal tumors has been well documented not only in the sporadic [7] but also in the colitis-associated model of CRC. [28],[92] However, Ishikawa et al. have recently demonstrated that cyclooxygenase-derived prostanoids are not major players in colitis-associated cancer. [93] Indeed, despite a significant elevation of COX-2 expression in AOM/DSS-induced colon tumors, similar tumors also developed in AOM/DSS-treated Cox-2 2/2− and Cox-1 2/2− knockout mice. On the other hand, tumor formation induced by multiple injections of AOM (with no DSS-induced colitis) did not occur in Cox-2 2/2− knockout mice. These data suggest that the factors responsible for tumor promotion in colitis-associated CRC differ from those that promote both inherited and sporadic CRC.
iNOS
Although the role of mast cells, which can enhance intestinal iNOS activity, in IBD development is still a matter of debate, it has been suggested that iNOS is involved in IBD and IBD-related colon carcinogenesis. [94]
It has been extensively demonstrated that all large bowel neoplasms as well as dysplasia that develop within 20 weeks after the start of the AOM/DSS treatment show positive reactivity for iNOS. [28] However, there has been lack of correlation between immunohistochemical signal/Western blot assay and iNOS mRNA level [82] found in tumors of ICR mice treated with AOM and DSS. [28],[92] One possible explanation for this discrepancy might be that iNOS overexpression is at least partly localized to tumor stroma, especially to tumor-associated macrophages. It is also possible that there is no simple relationship between steady-state transcript level and mature protein. Recent data from Seril et al. [95] indicate that mouse iNOS is not essential for the development of colitis-associated tumorigenesis, even though iNOS inhibition attenuates tumorigenesis in mice treated with DSS. [96]
Others
CAR1 (constitutive androstane receptor), a member of nuclear receptor superfamily, [97] may play an important role in the AOM/DSS carcinogenesis model as this protein is drastically decreased in AOM/DSS-induced tumors. [61] Specifically, CAR plays central roles in protecting the body against environmental xenobiotics. [98]
The comprehensive DNA microarray analysis using colonic mucosa of AOM/DSS-treated and AOM/DSS-untreated mice has revealed that certain CYP families (CYP2d26, CYP4f16, and CYP2c55) are downregulated in the colonic mucosa of mice that receive AOM/DSS, suggesting that CYP may play an important role in inflammation-related colon carcinogenesis. [62]
Inflammation and Tumorigenesis in The AOM/DSS Model
The multistep process of carcinogenesis is characterized by the canonical phases of initiation, promotion, and progression. The tumor-promotion process requires both sustained exposure to agents that stimulate growth, as well as inhibition of apoptosis of cells initiated by the earlier exposure to the carcinogen. The inflammatory microenvironment classically affects tumor promotion in its role as an altered stem cell niche but can also affect tumor initiation and tumor progression. [99] Mounting evidence from preclinical and clinical studies suggest that persistent inflammation is an essential component of all tumors, and some of the underlying molecular mechanisms have been elucidated.[100] The bacterium Helicobacter pylori, as an example, is one of the main contributing factors to the development of gastric cancer, the second most common cause of cancer-related mortality worldwide. [101]
Interestingly, tumors of the gastrointestinal tract provide a paradigmatic connection between inflammation and cancer. Epidemiological studies have shown that patients with IBD, including UC, are at increased risk of developing CRC. [102] IBD-associated colon carcinogenesis can be summarized as an inflammation-dysplasia-carcinoma sequence: hyperplastic lesions in the inflamed mucosa develop CRC after passing through the phase of flat dysplasia.
A number of possible mechanisms by which inflammation can contribute to carcinogenesis have been elucidated, including induction of genomic instability, alterations in epigenetic events and subsequent inappropriate gene expression, enhanced proliferation of initiated cells, resistance to apoptosis, aggressive tumor neovascularization, invasion through tumor-associated basement membrane and metastasis, the production of inflammation-induced reactive oxygen and nitrogen species. All of these mechanisms can directly or indirectly contribute to malignant cell transformation by causing damage to important cellular components (e.g., DNA, proteins, and lipids). In this context, several inflammatory mediators have emerged as key players in the initiation and development of CRC.
The AOM/DSS model represents a very valuable murine model to examine the role of inflammation in colon carcinogenesis, because it is a variation of a standard UC model, which typically requires long exposure periods or many cycles of DSS administration, before resulting in a relatively low tumor incidence. [103]
Even after only 3-4 weeks of treatment, signs of diffuse inflammation are present in the colonic mucosa of AOM/DSS-treated mice; epithelial crypts are distorted and irregularly distributed in the lamina propria, which contains high numbers of inflammatory cells, including lymphocytes and plasma cells. [28],[104] Interestingly, extensive mast cell migration can be found surrounding or within carcinomas.[28] This parallels what is observed in humans, where colitis-associated cancer is characterized by a dense infiltrate of immune cells, including macrophages, dendritic cells, and T-cells (which include the subset of CD8+ and CD4+ effector T-cells and CD4+ CD25+ T-cells). [105] Recently, Garrett et al. reported that dendritic cells were the necessary cellular effectors for the proinflammatory carcinogenic program of their T-Bet(−/−) RAG2(−/−) UC model. [106]
Importantly, during the first phase of tumor development in the AOM/DSS model it is possible to observe both conditions predisposing to cancer – colitis and genetic/epigenetic events – orchestrate the construction of a particular microenvironment which contributes to tumor progression. Indeed, alterations of genes representative of all classes of oncogenes drive the production of inflammatory mediators. Thus, an intrinsic pathway of inflammation (driven by genetic alteration in tumor cells leading to the expression of inflammation-related programs) as well as an extrinsic pathway (driven by inflammatory conditions in the tumor microenvironment) have been identified, both of which promote tumor growth. [107]
Key components of cancer-promoting inflammation have been investigated in the AOM/DSS model, including master transcription factors (e.g., nuclear factor kB and STAT3), proinflammatory cytokines (TNF-α, interleukin-6), COX-2, and selected chemokines. Of no less importance are mediators that keep inflammation in check, including IL-10, TGF-β, toll-like receptor (TLR), and the IL-1 receptor inhibitor TIR8 (also known as single immunoglobulin IL-1R-related molecule, SIGIRR), and the chemokine decoy and scavenger receptor D6.
Toll-like receptors
In general, a close link between innate and adaptive immunity, and inflammation and carcinogenesis is well established in many forms of cancer, including CRC. For example, evidence is mounting to support a role for TLRs in carcinogenesis. TLR4 is upregulated in intestinal epithelial cells in active CD and UC and its signaling induces COX-2, prostaglandin E2, and reactive oxygen species. [108] Using the AOM/DSS mouse model of CRC, it was shown that TLR4-deficient mice exhibited significantly reduced tumor number and size compared with wild-type controls. [109]
TNF-α
TNF-α is a key regulator of inflammation, inducing the recruitment of intracellular adaptor proteins. Its receptor (TNFR) can trigger NF-kB and downstream cell survival pathways as well as caspase-8 and associated apoptotic pathways. [110] TNF-α is found in the microenvironment of many types of tumor, including breast, ovarian, colorectal, prostate, melanoma, lymphoma, and leukemia. Specifically, in CRC, the mRNA and protein levels of TNF-α are increased to abnormal levels in the preneoplastic inflamed colonic mucosa. [111] The role of the signaling pathways downstream of TNF-α was explored in mice deficient in the type 1 TNFR, p55. [112] In the absence of TNF-α signaling, in response to the AOM/DSS challenge, there was decreased mucosal damage, inflammatory cell infiltrate, and cytokine expression in the mucosa, as well as a significant reduction in tumor formation. When the specific TNF-α antagonist etanercept was administered in the AOM-challenged mouse model, neutrophil and macrophage infiltration into the mucosa was decreased, as was tumor number and size. [113]
NF-kB
NF-kB (nuclear factor of kappa light-chain gene enhancer in B cells) is activated by signaling pathways mediated through TLRs and other microorganism-sensing molecules, or by TNF-α and IL-1β. [107] It mediates the transcription of downstream targets that can belong to four functional categories: antiapoptotic genes, inflammatory and immunoregulatory genes, genes that promote cell-cycle progression, and genes that encode negative regulators of NF-kB. Many of these targets have been associated with cancer initiation and progression. [114] Greten et al. [115] used the AOM/DSS model to show that when NF-kB activity was disrupted in colonic epithelial cells of AOM/DSS-treated mice, there was a dramatic reduction in tumor number. This was associated with enhanced epithelial cell apoptosis during early tumor development, but there was no reduction in inflammation. The authors thus demonstrated that NF-kB activation in epithelial cells contributes to tumor initiation and promotion, primarily by suppressing apoptosis.
Of interest is netrin-1, a soluble protein that has been proposed to play a crucial role during colorectal tumorigenesis by regulating apoptosis. [116] Together with the observation that the netrin-1 gene is a transcriptional target for NF-kB [117] and that IBD-associated CRC is linked to NF-kB and survival [116] Paradisi et al. demonstrated that CRCs from IBD patients have selected upregulation of netrin-1 and that overexpression of netrin-1 in AOM/DSS-treated mice in the gastrointestinal tract is associated with IBD-associated CRC. [117]
JAK/STAT pathway
Finally, the AOM/DSS model has also been used to demonstrate the importance of the JAK/STAT pathway (Janus kinase/signal transducers and activators of transcription).[118] Indeed loss of Socs1 and Socs3 (suppressors of cytokine signaling) expression results in increased activation of STAT1, STAT3, and NF-kB and the development of colorectal tumors.[119],[120] A direct link between Socs signaling and Myc exists since AOM/DSS-induced adenocarcinomas from Socs1-deficient mice have increased levels of nuclear β-catenin and Myc expression when compared to tumors of Socs1 wild-type mice. [119] Intestinal epithelium deficient for Socs3 does not result in chronic inflammation or development of tumors. However, when treated with AOM/DSS, these mice develop colon tumors that is preceded by inflammation, suggesting that Socs3 expression in neighboring stroma may be required to suppress chronic inflammation and, subsequently, tumor promotion.
Epigenetic Modification Arising in The AOM/DSS Mode
Environmental and genetic factors contribute to CRC formation by promoting the accumulation of gene mutations and epigenetic alterations in colon epithelial cells that drive the polyp-cancer formation process. Although aberrant DNA methylation, one of the most common epigenetic alterations, has been recently shown to occur in CRC, the causal role of these epigenetic changes in the process of cancer initiation and promotion is poorly understood at this time. It has been established that aberrant hypermethylation of tumor suppressor genes can result in their transcriptional silencing, which is the mechanism through which DNA methylation is believed to promote cancer formation. DNA methylation appears to cooperate with concurrent alterations in chromatin structure to repress transcription. [121] However, little is known regarding the precise timing of these epigenetic alterations in the transition of normal colon epithelial cells to cancer cells through the polyp-cancer progression sequence. Furthermore, the biological role of these aberrantly methylated genes in the development of CRC is still poorly understood.
Recent studies have suggested that alterations in DNA methylation can contribute to tumor pathogenesis in mouse models similarly to what happens in human cancer. It has indeed been shown in both the AOM-induced and mutant Apc mouse models of intestinal cancer that the global DNA hypomethylation observed in human colorectal malignancy [122] is also present in tumors arising in these mice. [60],[123]
Furthermore, the AOM colon cancer model (lacking the initial colitis induction) has been used for assessing the methylation state of tumor suppressor genes to determine the role of epigenetic alterations in cancer initiation and progression. Borinstein et al. [124] investigated the methylation status of 11 candidate genes known to be aberrantly methylated in human cancer or mouse tumor models and demonstrated the presence of specific methylation patterns in tumors and normal tissues. Of note, two of these genes, Zik1 (zinc finger protein-interacting with K protein 1) and Gja9 (gap junction protein alpha-9), show aberrant DNA methylation in the tumors, which is either not present or is present at low frequency in the normal colon. The aberrant DNA methylation of Gja9 has not been reported previously, instead Zik1 has also been identified as being hypermethylated in intestinal metaplasia (IM) of the stomach, in gastric cancer cell lines, and in primary gastric cancer tumor samples, suggesting that epigenetic silencing of Zik1 may contribute to the pathogenesis of gastric neoplasia and may play an important role in the transformation of gastrointestinal mucosa from normal to cancerous. [125]
Recent studies suggest that alterations in DNA methylation can contribute to tumor pathogenesis in mouse models, indicating that AOM-induced mouse colonic tumors can recapitulate human CRC, although the frequency of aberrantly methylated genes in the mouse model appears to be less common than in human CRC. Further studies of the epigenetic modifications using genome-wide assays should provide a comprehensive assessment of the methylation state of the DNA in colon cancers arising in AOM-based CRC murine models in order to highlight the potential of these models to study a range of issues that remains to be resolved in the field of human epigenetic aberrations.
Implications for Chemoprevention – The AOM/DSS Model In Preclinical Studies
Although the number of chemoprevention studies reported in the AOM/DSS model is quite limited, recent and emerging data show that colitis-associated colorectal carcinogenesis could be considered a preventable disease. Preclinical studies conducted to date support the use of the AOM/DSS mouse model as a highly relevant system that could be considered a valuable in vivo platform for chemopreventive intervention studies.
The following section summarizes the ability of selected classes of both synthetic and natural dietary agents to inhibit the formation of AOM/DSS-induced neoplasms [Table 1].
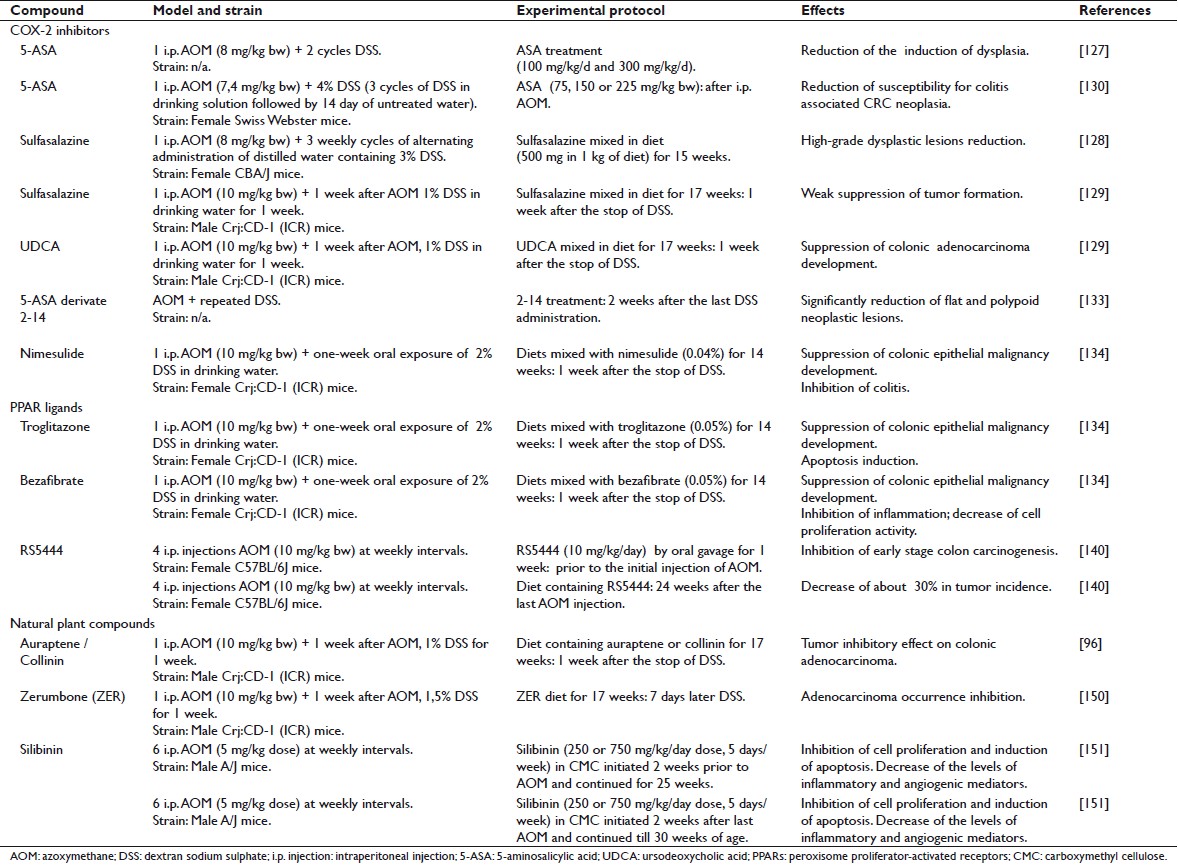 |
Table 1: Chemopreventive preclinical studies performed in the AOM/DSS model Click here to view |
Cox-2 inhibitors
Although the significant contribution of COX-2 to the development of sporadic colorectal tumors has been well documented in both preclinical [7] and clinical [126] studies, the effect of the anti-inflammatory agent 5-aminosalicylic acid (5-ASA) on the risk for colitis-associated CRC is still controversial. The effect of 5-ASA and its derivates on colorectal carcinogenesis has been evaluated in the AOM/DSS mouse model. Grimm et al. [127] studied the chemopreventive effects of low (100 mg/kg) and high (300 mg/kg) doses of 5-ASA in the AOM/DSS model. In this study, mice underwent two cycles of DSS treatment after a single i.p. injection of AOM (8 mg/kg).
Similarly, the administration of sulfasalazine, a derivative of 5-ASA, to AOM/DSS-treated mice reduced the number of areas of high-grade dysplasia to less than 20% compared to the AOM/DSS controls. [128] In this study only a single dose of sulfasalazine was evaluated, but the effect of this agent on morphological subtypes of lesions and inflammation was not examined. Kohno et al. [129] reported that the suppressive effects of sulfasalazine on CRC were relatively weak when the drug was given in the promotion/progression phase of the development of the disease. This discrepancy may be due to differences in experimental protocols and the murine strains used. Kohno et al. also reported that treatment with ursodeoxycholic acid (UDCA) resulted in inhibition of AOM/DSS-induced CRC in mice, without causing any adverse effects.
A study conducted by Clapper et al. [130] evaluated the ability of 5-ASA to inhibit colitis-associated colorectal neoplasia in the model. The results strongly suggest that chronic exposure to 5-ASA is able to reduce susceptibility to colitis-associated CRC. Although most of the beneficial effects of 5-ASA have been attributed to its ability to inhibit cyclooxygenases and prostaglandin H synthase, in this study there was no significant difference in the level of expression of COX-2 (stromal cells or epithelial cells) within the colons of AOM/DSS control mice as compared to those treated with 5-ASA, suggesting that the chemopreventive activity of 5-ASA is not related to the expression level of COX-2. Indeed, in a previous study, [131] 5-ASA has been shown to inhibit the proliferation of DLD1 colon carcinoma cells, which are deficient in cyclooxygenase.
Weber et al. [132] have shown the ability of 5-ASA to suppress NF-kB activation. Decreased binding of NF-kB to target genes leads to a reduction in the expression of proinflammatory cytokines, adhesion molecules, cytokines, inflammatory mediators, and antiapoptotic genes.
A novel 5-ASA derivative called ‘5-ASA derivative 2-14’ seems to be 10-fold more potent than 5-ASA in inhibiting CRC cell proliferation. [133] Proliferating cell nuclear antigen (PCNA) staining confirmed the antiproliferative effect of 2-14. By contrast, there was no significant change in PCNA staining in the normal colonic mucosa of mice treated with 2-14, confirming that this compound does not substantially affect the growth of normal colonic cells.
Besides 5-ASA and its derivatives, selective COX-2 inhibitors have been tested in the colorectal carcinogenesis animal model. Particularly, nimesulide (4-nitro-2-phenoxymethanesulfonanilide), a selective inhibitor of COX-2, effectively inhibited AOM/DSS-induced colitis-related colonic carcinogenesis in mice. [134] The suppressive effects of nimesulide on the development of colonic adenocarcinoma correlates well with the inhibition of cell proliferation activity, induction of apoptosis, and the lower immunoreactivity of β-catenin, COX-2, iNOS, and nitrotyrosine in colonic malignancies. These data suggest that nimesulide could be applied as an effective chemopreventor of both sporadic and IBD-associated colorectal carcinogenesis.
Peroxisome proliferator-activated receptor ligands
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors, belonging to the nuclear hormone receptor superfamily. [135] Three PPAR isotypes, PPARα, PPARβ, and PPARγ, have been identified. PPARγ is highly expressed in fat tissue and plays an important role in adipocyte differentiation and lipid storage. It is also expressed in a number of epithelial neoplasms, such as cancers in colon, breast, and prostate. PPARγ ligands, such as troglitazone and rosiglitazone can induce apoptosis and adipogenic differentiation and inhibit tumor growth both in vitro and in vivo. [136],[137] In different studies conducted by Tanaka et al., [129],[138] the chemopreventive ability of the PPARγ ligand troglitazone or the PPARα ligand bezafibrate has been characterized in the AOM/DSS-induced mouse colon carcinogenesis model. Inhibition of inflammation in colon mucosa and decrease in cell proliferation activity caused by these PPAR ligands might be responsible for their chemopreventive effects in colitis-associated colon carcinogenesis. [139] A new third-generation thiazolidinedione, RS5444, which is about 50 times more potent than rosiglitazone, was tested in the AOM/DSS mouse model. [140],[141] RS5444 is able to significantly suppress ACF formation in AOM-treated mice [142] and may therefore have chemopreventive efficacy. PPARγ ligands inhibited proliferation of normal colonic epithelial cells but they did not show statistically significant effects on adenomatous epithelial cells, suggesting that the ability of these compounds to inhibit colonic epithelial cell proliferation is lost at an early stage in carcinogenesis. Moreover RS5444-treated mice have shown a decrease of about 30% in tumor incidence, though this reduction was not statistically significant. In this study, tumor volume was highly variable and no significant effect on tumor size was observed. Although RS5444 did not inhibit the growth of established tumors, it was observed that control tumors were significantly more dysplastic than the tumors from RS5444-treated mice. It seems indeed that PPARγ ligands promote differentiation of both normal and transformed gastrointestinal epithelial cells, [143] consistently with their ability to promote differentiation of minimally transformed colonic epithelial cells. The ability to block initiation of transformation may also be explained by their ability to inhibit the proliferation of normal colonic epithelial cells, thereby decreasing the numbers of target cells where the carcinogen could act.
Natural plant compounds
Epidemiological studies clearly demonstrate the existence of an inverse correlation between a diet rich in fruits/vegetables and human colon cancer. Fruits and vegetables contain active compounds able to reduce the risk of this tumor. Several natural and semisynthetic agents with anti-inflammatory properties have been reported to have chemopreventive activity against carcinogenesis in preclinical animal studies. Prenyloxycoumarins, including auraptene and collinin, are secondary metabolites that are mainly found in plants belonging to the families Rutaceae and Umbelellierae (orange, lemon, grapefruit, etc). Auraptene and collinin are reported to have anti-inflammatory activity. [144] Auraptene significantly attenuates the lipopolysaccharide (LPS)-induced protein expression of iNOS and COX-2, as well as the release of TNF-α. [145] Auraptene and collinin are also able to inhibit platelet aggregation induced by arachidonic acid and platelet activating factor in vitro.[146]
The effects of these natural compounds have been examined in AOM/DSS mouse colon carcinogenesis model. [96] The incidence of colorectal adenocarcinomas in AOM/DSS mouse with different dosages (0.01% and 0.05%) of auraptene and collinin was significantly smaller than that in the untreated group. The multiplicity of colon adenocarcinomas and colonic inflammation scores in those groups was also significantly lower than that of the control group. The suppressive effect of auraptene and collinin on the development of colonic adenocarcinoma correlated with the inhibition of cell proliferation activity (PCNA-labeling index of colonic adenocarcinoma), induction of apoptosis, and inhibition of immunoreactivity of COX-2 and iNOS in the colonic malignancies. The protective effects of dietary auraptene and collinin after treatment with AOM and DSS might be mainly due to their ability to inhibit inflammation and oxidative stress. [96] Dietary administration of the prenyloxycoumarins, auraptene and collinin, could effectively suppress colitis-related colon carcinogenesis induced by AOM and DSS.
The chemopreventive property of auraptene and a novel prodrug, 4′-geranyloxy-ferulic acid (GOFA), has been tested using inclusion complexes with beta-cyclodextrin (β-CD): GOFA/β-CD and AUR/β-CD. These compounds exert their chemopreventive effect by modulating cell proliferation, inducing apoptosis, and suppressing proinflammatory cytokines (NF-kB, Nrf2, TNF-α, Stat3, IL-6, and IL-1β) in adenocarcinomas that develop in the setting of colitis. In turn, the expression of these cytokines may be involved in AOM/DSS-induced colon tumorigenesis. It has been demonstrated that treatment with GOFA/β-CD and AUR/β-CD significantly lowers colonic inflammation induced by DSS. Therefore, the suppression of chronic inflammation through the modulation of expression of several proinflammatory gene products that mediate several events of carcinogenesis may be responsible for the chemopreventive properties of these compounds. [147]
Recent works have elucidated several biological properties of zerumbone (ZER), a major constituent of the subtropical ginger plant, Zingiber zerumbet Smith, which seems to inhibit carcinogenesis through inhibition of free radical generation, iNOS and COX-2 expression, and TNF-α release in activated leukocytes, as well as induction of apoptosis in human colonic adenocarcinoma cell lines. [148] Dietary feeding with ZER strongly suppresses DSS-induced acute colitis in mice [149] and the formation of aberrant crypt foci (ACF).
The incidence and the multiplicity of colonic adenocarcinoma in mice groups receiving ZER at different doses (100 ppm, 250 ppm, and 500 ppm) have been demonstrated to be less than that in AOM/DSS-treated mice not receiving the natural compound. ZER feeding significantly decreased the PCNA labeling index [150] and lowered the immunohistochemical scores of NF-kB and HO-1 (heme oxygenase), the expression of which correlates with carcinogenesis and inflammation.
Silibinin, a flavanolignan isolated from Silybum marianum, has been tested in the AOM mouse model. Interestingly, both low (250 mg/kg) and high (750 mg/kg) doses of silibinin, besides inhibiting cell proliferation and inducing apoptosis, impacts the levels of inflammatory and angiogenic mediators such as COX-2, iNOS, and VEGF, suggesting that silibinin has chemopreventive efficacy in colon tumorigenesis due to its effects on two key events of colon carcinogenesis: inflammation and angiogenesis. [151]
Different preclinical studies support the use of the AOM/DSS-induced colitis mouse model as a highly relevant system that allows characterization of the molecular events required for the formation of colorectal neoplasia in the setting of chronic inflammation. Results from chemopreventive intervention studies provide evidence that colitis-associated CRC can be prevented.
Conclusions and Future Perspectives
In the last two decades murine models of CRC have provided detailed insight into the histopathologic and molecular processes underlying colorectal tumor progression and have contributed to our understanding of human CRC as well as IBD-related CRC pathogenesis.
As powerful model systems, the chemically-induced CRC murine models have proved highly valuable. Among them the AOM/DSS murine model has been described to be a reliably practical tool: tumor development is predictable and consistent, with a high incidence of affected animals in a narrow time frame. For its particular ability in reproducing many aspects of human CRC, along with the possibility it offers for studying the earliest succession of events underlying tumor initiation when single aberrant crypts appear in colonic mucosa, the AOM/DSS model is a powerful experimental platform that will be very useful in the development of novel and effective chemopreventive agents against CRC; it is also a valuable model for investigating novel diagnostic, prognostic, and predictive markers for use in clinical practice.
In the future, further improvement of the AOM/DSS model, in combination with the development of novel and more sophisticated molecular tools and genetic strategies, might enable us to define new aspects of CRCs, for example dissecting through high resolution approaches the molecular networks alterated in the earliest phase of this neoplastic disease. It should aid in the identification of the genetic and epigenetic events, still partially unknown, that act in the preneoplastic phase of colon carcinogenesis and which are fundamental in CRC development.
Acknowledgments
This review was based on studies supported in part by Project ‘FILAS-DTB 2008,’ ‘FIRB 2006 RBIP0695BB,’ Project ‘Fondazione Roma, 2009,’ and grant ‘AIRC MFAG 10520’.
References
| 1. | van Hogezand RA, Eichhorn RF, Choudry A, Veenendaal RA, Lamers CB. Malignancies in inflammatory bowel disease: fact or fiction? Scand J Gastroenterol Suppl 2002;236:48-53.  |
| 2. | Powell SM, Petersen GM, Krush AJ, Booker S, Jen J, Giardiello FM, et al. Molecular diagnosis of familial adenomatous polyposis. N Engl J Med 1993;329:1982-7. |
| 3. | Marian B. Colorectal cancer: modeling causes, prevention and therapy. Drug Discov Today Dis Mod 2004;1:1-7. |
| 4. | Friedman E, Urmacher C, Winawer S. A model for human colon carcinoma evolution based on the differential response of cultured preneoplastic, premalignant, and malignant cells to 12-O-tetradecanoylphorbol-13-acetate. Cancer Res 1984;44:1568-78. |
| 5. | Fodde R, Smits R. Disease model: familial adenomatous polyposis. Trends Mol Med 2001;7:369-73. |
| 6. | Takaku K, Sonoshita M, Sasaki N, Uozumi N, Doi Y, Shimizu T, et al. Suppression of intestinal polyposis in Apc(delta 716) knockout mice by an additional mutation in the cytosolic phospholipase A(2) gene. J Biol Chem 2000;275:34013-6. |
| 7. | Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996;87:803-9. |
| 8. | Seno H, Oshima M, Ishikawa TO, Oshima H, Takaku K, Chiba T, et al. Cyclooxygenase 2- and prostaglandin E(2) receptor EP(2)-dependent angiogenesis in Apc(Delta716) mouse intestinal polyps. Cancer Res 2002;62:506-11. |
| 9. | Heyer J, Yang K, Lipkin M, Edelmann W, Kucherlapati R. Mouse models for colorectal cancer. Oncogene 1999;18:5325-33. |
| 10. | Corpet DE, Pierre F. Point: From animal models to prevention of colon cancer. Systematic review of chemoprevention in min mice and choice of the model system. Cancer Epidemiol Biomarkers Prev 2003;12:391-400. |
| 11. | Gebert HF, Jagelman DG, McGannon E. Familial polyposis coli. Am Fam Physician 1986,33:127-37. |
| 12. | Garofalo A, Chirivi RG, Scanziani E, Mayo JG, Vecchi A, Giavazzi R. Comparative study on the metastatic behavior of human tumors in nude, beige/nude/xid and severe combined immunodeficient mice. Invasion Metastasis 1993;13:82-91. |
| 13. | Hoffman RM. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: a bridge to the clinic. Invest New Drugs 1999;17:343-59. |
| 14. | Hasegawa R, Sano M, Tamano S, Imaida K, Shirai T, Nagao M, et al. Dose-dependence of 2-amino-1-methyl-6-phenylimidazo[4,5-b]-pyridine (PhIP) carcinogenicity in rats. Carcinogenesis 1993;14:2553-7. |
| 15. | Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis 2009;30:183-96. |
| 16. | Reddy BS, Rivenson A. Inhibitory effect of Bifidobacterium longum on colon, mammary, and liver carcinogenesis induced by 2-amino-3-methylimidazo[4,5-f]quinoline, a food mutagen. Cancer Res 1993;53:3914-8. |
| 17. | Doi K, Wanibuchi H, Salim EI, Morimura K, Kinoshita A, Kudoh S, et al. Lack of large intestinal carcinogenicity of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine at low doses in rats initiated with azoxymethane. Int J Cancer 2005;115:870-8. |
| 18. | Kobaek-Larsen M, Thorup I, Diederichsen A, Fenger C, Hoitinga MR. Review of colorectal cancer and its metastases in rodent models: comparative aspects with those in humans. Comp Med 2000;50:16-26. |
| 19. | Walpole A, Williams MH, Roberts DC. The carcinogenic action of 4-aminodiphenyl and 3:2′-dimethyl-4-amino-diphenyl. Br J Ind Med 1952;9:255-63. |
| 20. | Reddy BS, Ohmori T. Effect of intestinal microflora and dietary fat on 3,2′-dimethyl-4-aminobiphenyl-induced colon carcinogenesis in F344 rats. Cancer Res 1981;41:1363-7. |
| 21. | Narisawa T, Magadia NE, Weisburger JH, Wynder EL. Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N’-nitro-N-nitrosoguanidine in rats. J Natl Cancer Inst 1974;53:1093-7. |
| 22. | Laqueur GL. Carcinogenic effects of cycad meal and cycasin, methylazoxymethanol glycoside, in rats and effects of cycasin in germfree rats. Fed Proc 1964;23:1386-8. |
| 23. | Delker DA, McKnight SJ 3rd, Rosenberg DW. The role of alcohol dehydrogenase in the metabolism of the colon carcinogen methylazoxymethanol. Toxicol Sci 1998;45:66-71. |
| 24. | Sohn OS, Fiala ES, Requeijo SP, Weisburger JH, Gonzalez FJ. Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res 2001;61:8435-40. |
| 25. | Haase P, Cowen DM, Knowles JC. Histogenesis of colonic tumours in mice induced by dimethyl hydrazine. J Pathol 1973. |
| 26. | Deschner EE, Long FC. Colonic neoplasms in mice produced with six injections of 1,2-dimethylhydrazine. Oncology 1977;34:255-7. |
| 27. | Thurnherr N, Deschner EE, Stonehill EH, Lipkin M. Induction of adenocarcinomas of the colon in mice by weekly injections of 1,2-dimethylhydrazine. Cancer Res 1973;33:940-5. |
| 28. | Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci 2003;94:965-73. |
| 29. | Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc 2007;2:1998-2004. |
| 30. | Suzuki R, Kohno H, Sugie S, Nakagama H, Tanaka T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis 2006;27:162-9. |
| 31. | Bird RP, Good CK. The significance of aberrant crypt foci in understanding the pathogenesis of colon cancer. Toxicol Lett 2000;112:395-402. |
| 32. | Maskens AP. Histogenesis and growth pattern of 1,2-dimethylhydrazine-induced rat colon adenocarcinoma. Cancer Res 1976;36:1585-92. |
| 33. | Clapp NK, Henke MA, London JF, Shock TL. Enhancement of 1,2-dimethylhydrazine-induced large bowel tumorigenesis in Balb/c mice by corn, soybean, and wheat brans. Nutr Cancer 1984;6:77-85. |
| 34. | Jackson PE, Cooper DP, O’Connor PJ, Povey AC. The relationship between 1,2-dimethylhydrazine dose and the induction of colon tumours: tumour development in female SWR mice does not require a K-ras mutational event. Carcinogenesis 1999;20:509-13. |
| 35. | Hata K, Yamada Y, Kuno T, Hirose Y, Hara A, Qiang SH, et al. Tumor formation is correlated with expression of beta-catenin-accumulated crypts in azoxymethane-induced colon carcinogenesis in mice. Cancer Sci 2004;95:316-20. |
| 36. | Papanikolaou A, Wang QS, Papanikolaou D, Whiteley HE, Rosenberg DW. Sequential and morphological analyses of aberrant crypt foci formation in mice of differing susceptibility to azoxymethane-induced colon carcinogenesis. Carcinogenesis 2000;21:1567-72. |
| 37. | Bissahoyo A, Pearsall RS, Hanlon K, Amann V, Hicks D, Godfrey VL, et al. Azoxymethane is a genetic background-dependent colorectal tumor initiator and promoter in mice: effects of dose, route, and diet. Toxicol Sci 2005;88:340-5. |
| 38. | Okayasu I, Ohkusa T, Kajiura K, Kanno J, Sakamoto S. Promotion of colorectal neoplasia in experimental murine ulcerative colitis. Gut 1996;39:87-92. |
| 39. | Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004;21:491-501. |
| 40. | Becker C, Fantini MC, Wirtz S, Nikolaev A, Kiesslich R, Lehr HA, et al. In vivo imaging of colitis and colon cancer development in mice using high resolution chromoendoscopy. Gut 2005;54:950-4. |
| 41. | Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest 2008;118:2516-25. |
| 42. | Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 2003;124:762-77. |
| 43. | Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996;87:159-70. |
| 44. | Roncucci L, Medline A, Bruce WR. Classification of aberrant crypt foci and microadenomas in human colon. Cancer Epidemiol Biomarkers Prev 1991;1:57-60. |
| 45. | Ward JM. Morphogenesis of chemically induced neoplasms of the colon and small intestine in rats. Lab Invest 1974;30:505-13. |
| 46. | Riddell RH, Goldman H, Ransohoff DF, Appelman HD, Fenoglio CM, Haggitt RC, et al. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum Pathol 1983;14:931-68. |
| 47. | Pascal RR. Dysplasia and early carcinoma in inflammatory bowel disease and colorectal adenomas. Hum Pathol 1994;25:1160-71. |
| 48. | Cheng L, Lai MD. Aberrant crypt foci as microscopic precursors of colorectal cancer. World J Gastroenterol 2003;9:2642-9. |
| 49. | Roncucci L, Stamp D, Medline A, Cullen JB, Bruce WR. Identification and quantification of aberrant crypt foci and microadenomas in the human colon. Hum Pathol 1991;22:287-94. |
| 50. | Pretlow TP, Barrow BJ, Ashton WS, O’Riordan MA, Pretlow TG, Jurcisek JA, et al. Aberrant crypts: putative preneoplastic foci in human colonic mucosa. Cancer Res 1991;51:1564-7. |
| 51. | Nucci MR, Robinson CR, Longo P, Campbell P, Hamilton SR. Phenotypic and genotypic characteristics of aberrant crypt foci in human colorectal mucosa. Hum Pathol 1997;28:1396-407. |
| 52. | Takahashi M, Fukuda K, Sugimura T, Wakabayashi K. Beta-catenin is frequently mutated and demonstrates altered cellular location in azoxymethane-induced rat colon tumors. Cancer Res 1998;58:42-6. |
| 53. | Yamada Y, Yoshimi N, Hirose Y, Kawabata K, Matsunaga K, Shimizu M, et al. Frequent beta-catenin gene mutations and accumulations of the protein in the putative preneoplastic lesions lacking macroscopic aberrant crypt foci appearance, in rat colon carcinogenesis. Cancer Res 2000;60:3323-7. |
| 54. | Yamada Y, Yoshimi N, Hirose Y, Matsunaga K, Katayama M, Sakata K, et al. Sequential analysis of morphological and biological properties of beta-catenin-accumulated crypts, provable premalignant lesions independent of aberrant crypt foci in rat colon carcinogenesis. Cancer Res 2001;61:1874-8. |
| 55. | Caderni G, Femia AP, Giannini A, Favuzza A, Luceri C, Salvadori M, et al. Identification of mucin-depleted foci in the unsectioned colon of azoxymethane-treated rats: correlation with carcinogenesis. Cancer Res 2003;63:2388-92. |
| 56. | Paulsen JE, Løberg EM, Olstørn HB, Knutsen H, Steffensen IL, Alexander J. Flat dysplastic aberrant crypt foci are related to tumorigenesis in the colon of azoxymethane-treated rat. Cancer Res 2005;65:121-9. |
| 57. | Maltzman T, Whittington J, Driggers L, Stephens J, Ahnen D. AOM-induced mouse colon tumors do not express full-length APC protein. Carcinogenesis 1997;18:2435-9. |
| 58. | Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. Frequent mutations of the beta-catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis 2000;21:1117-20. |
| 59. | Wang QS, Papanikolaou A, Sabourin CL, Rosenberg DW. Altered expression of cyclin D1 and cyclin-dependent kinase 4 in azoxymethane-induced mouse colon tumorigenesis. Carcinogenesis 1998;19:2001-6. |
| 60. | Vivona AA, Shpitz B, Medline A, Bruce WR, Hay K, Ward MA, et al. K-ras mutations in aberrant crypt foci, adenomas and adenocarcinomas during azoxymethane-induced colon carcinogenesis. Carcinogenesis 1993;14:1777-81. |
| 61. | Yasui Y, Tanaka T. Protein expression analysis of inflammation-related colon carcinogenesis. J Carcinog 2009;8:10. [PUBMED]  |
| 62. | Suzuki R, Miyamoto S, Yasui Y, Sugie S, Tanaka T. Global gene expression analysis of the mouse colonic mucosa treated with azoxymethane and dextran sodium sulfate. BMC Cancer 2007;7:84. |
| 63. | Miller JR, Moon RT. Signal transduction through beta-catenin and specification of cell fate during embryogenesis. Genes Dev 1996;10:2527-39. |
| 64. | Takahashi M, Mutoh M, Kawamori T, Sugimura T, Wakabayashi K. Altered expression of beta-catenin, inducible nitric oxide synthase and cyclooxygenase-2 in azoxymethane-induced rat colon carcinogenesis. Carcinogenesis 2000,21:1319-27. |
| 65. | Cooper HS, Murthy S, Kido K, Yoshitake H, Flanigan A. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis 2000;21:757-68. |
| 66. | Mikami T, Mitomi H, Hara A, Yanagisawa N, Yoshida T, Tsuruta O, et al. Decreased expression of CD44, alpha-catenin, and deleted colon carcinoma and altered expression of beta-catenin in ulcerative colitis-associated dysplasia and carcinoma, as compared with sporadic colon neoplasms. Cancer 2000;89:733-40. |
| 67. | Tanaka T, Suzuki R, Kohno H, Sugie S, Takahashi M, Wakabayashi K. Colonic adenocarcinomas rapidly induced by the combined treatment with 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and dextran sodium sulfate in male ICR mice possess beta-catenin gene mutations and increases immunoreactivity for beta-catenin, cyclooxygenase-2 and inducible nitric oxide synthase. Carcinogenesis 2005;26:229-38. |
| 68. | Kohno H, Suzuki R, Sugie S, Tanaka T. Beta-Catenin mutations in a mouse model of inflammation-related colon carcinogenesis induced by 1,2-dimethylhydrazine and dextran sodium sulfate. Cancer Sci 2005;96:69-76. |
| 69. | Koesters R, Hans MA, Benner A, Prosst R, Boehm J, Gahlen J, et al. Predominant mutation of codon 41 of the beta-catenin proto-oncogene in rat colon tumors induced by 1,2-dimethylhydrazine using a complete carcinogenic protocol. Carcinogenesis 2001;22:1885-90. |
| 70. | Svec J, Ergang P, Mandys V, Kment M, Pácha J. Expression profiles of proliferative and antiapoptotic genes in sporadic and colitis-related mouse colon cancer models. Int J Exp Path 2010;91:44-53. |
| 71. | Valencia A, Chardin P, Wittinghofer A, Sander C. The ras protein family: evolutionary tree and role of conserved amino acids. Biochemistry 1991;30:4637-48. |
| 72. | Takahashi M, Wakabayashi K. Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci 2004;95:475-80. |
| 73. | Jacoby RF, Llor X, Teng BB, Davidson NO, Brasitus TA. Mutations in the K-ras oncogene induced by 1,2-dimethylhydrazine in preneoplastic and neoplastic rat colonic mucosa. J Clin Invest 1991;87:624-30. |
| 74. | Takesue F, Korenaga D, Yao T, Kabashima A, Sugimachi K. Development of colonic neoplasms and expressions of p53 and p21 proteins of experimental colitis of mice induced by dextran sulfate sodium. J Exp Clin Cancer Res 2001;20:413-8. |
| 75. | Suzui M, Yoshimi N, Ushijima T, Hirose Y, Makita H, Wang A, et al. No involvement of Ki-ras or p53 gene mutations in colitis-associated rat colon tumors induced by 1-hydroxyanthraquinone and methylazoxymethanol acetate. Mol Carcinog 1995;12:193-7. |
| 76. | Nambiar PR, Giardina C, Guda K, Aizu W, Raja R, Rosenberg DW. Role of the alternating reading frame (P19)-p53 pathway in an in vivo murine colon tumor model. Cancer Res 2002;62:3667-74. |
| 77. | Minamoto T, Buschmann T, Habelhah H, Matusevich E, Tahara H, Boerresen-Dale AL, et al. Distinct pattern of p53 phosphorylation in human tumors. Oncogene 2001;20:3341-7. |
| 78. | Knoepfler PS. Myc goes global: new tricks for an old oncogene. Cancer Res 2007;67:5061-3. |
| 79. | Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang XF. Transforming growth factor beta-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol Cell Biol 2004;24:2546-59. |
| 80. | Yekkala K, Baudino TA. Inhibition of intestinal polyposis with reduced angiogenesis in ApcMin/+ mice due to decreases in c-Myc expression. Mol Cancer Res 2007,5:1296-303. |
| 81. | Zhang X, Ge YL, Tian RH. The knockdown of c-myc expression by RNAi inhibits cell proliferation in human colon cancer HT-29 cells in vitro and in vivo. Cell Mol Biol Lett 2009;4:305-18. |
| 82. | Choi J, Southworth LK, Sarin KY, Venteicher AS, Ma W, Chang W, et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet 2008;4:e10. |
| 83. | Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta 2008;1782:197-228. |
| 84. | Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA 1993;90:770-4. |
| 85. | Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res 1999;59:3379-86. |
| 86. | Abdelaziz N, Colombo F, Mercier I, Calderone A. Nitric oxide attenuates the expression of transforming growth factor-beta(3) mRNA in rat cardiac fibroblasts via destabilization. Hypertension 2001;38:261-6.th |
| 87. | Becker C, Fantini MC, Neurath MF. TGF-beta as a T cell regulator in colitis and colon cancer. Cytokine Growth Factor Rev 2006;17:97-106. |
| 88. | Fantini MC, Rizzo A, Fina D, Caruso R, Sarra M, Stolfi C, et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology 2009;136:1308-16, e1-3. |
| 89. | Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994;107:1183-8. |
| 90. | Sinicrope FA, Gill S. Role of cyclooxygenase-2 in colorectal cancer. Cancer Metastasis Rev 2004;23:63-75. |
| 91. | Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell 1995;83:493-501. |
| 92. | Kim HS, Kundu JK, Le JS, Oh TY, Na HK, Surh YJ. Chemopreventive effects of the standardized extract (DA-9601) of Artemisia asiatica on azoxymethane-initiated and dextran sulfate sodium-promoted mouse colon carcinogenesis. Nutr Cancer 2008;60:90-7. |
| 93. | Ishikawa TO, Herschman HR. Tumor formation in a mouse model of colitis-associated colon cancer does not require COX-1 or COX-2 expression. Carcinogenesis 2010;31:729-36. |
| 94. | Chichlowski M, Westwood GS, Abraham SN, Hale LP. Role of mast cells in inflammatory bowel disease and inflammation-associated colorectal neoplasia in IL-10-deficient mice. PLoS One 2010;5:e12220. |
| 95. | Seril DN, Liao J, Yang GY. Colorectal carcinoma development in inducible nitric oxide synthase-deficient mice with dextran sulfate sodium-induced ulcerative colitis. Mol Carcinog 2007;46:341-53. |
| 96. | Kohno H, Suzuki R, Curini M, Epifano F, Maltese F, Gonzales SP, et al. Dietary administration with prenyloxycoumarins, auraptene and collinin, inhibits colitis-related colon carcinogenesis in mice. Int J Cancer 2006;118:2936-42. |
| 97. | Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science 2001;294:1866-70. |
| 98. | Maglich JM, Stoltz CM, Goodwin B, Hawkins-Brown D, Moore JT, Kliewer SA. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol Pharmacol 2002;62:638-46. |
| 99. | Quante M, Wang TC. Inflammation and stem cells in gastrointestinal carcinogenesis. Physiology (Bethesda) 2008;23:350-9. |
| 100. | Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140:883-99. |
| 101. | Houghton J, Wang TC. Helicobacter pylori and gastric cancer: a new paradigm for inflammation-associated epithelial cancers. Gastroenterology 2005;128:1567-78. |
| 102. | Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol 2004;287:G7-17. |
| 103. | Okayasu I, Yamada M, Mikami T, Yoshida T, Kanno J, Ohkusa T. Dysplasia and carcinoma development in a repeated dextran sulfate sodium-induced colitis model. J Gastroenterol Hepatol 2002;17:1078-83. |
| 104. | Paradisi A, Maisse C, Coissieux MM, Gadot N, Lépinasse F, Delloye-Bourgeois C, et al. Netrin-1 up-regulation in inflammatory bowel diseases is required for colorectal cancer progression. Proc Natl Acad Sci USA 2009;106:17146-51. |
| 105. | Waldner M, Schimanski CC, Neurath MF. Colon cancer and the immune system: the role of tumor invading T cells. World J Gastroenterol 2006;12:7233-8. |
| 106. | Garrett WS, Punit S, Gallini CA, Michaud M, Zhang D, Sigrist KS, et al. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell 2009;16:208-19. |
| 107. | Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008;454:436-44. |
| 108. | Fukata M, Abreu MT. Role of Toll-like receptors in gastrointestinal malignancies. Oncogene 2008;27:234-43. |
| 109. | Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007;133:1869-81. |
| 110. | Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev 2006;25:409-16. |
| 111. | Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer 2009;9:361-71. |
| 112. | Noguchi M, Hiwatashi N, Liu Z, Toyota T. Secretion imbalance between tumour necrosis factor and its inhibitor in inflammatory bowel disease. Gut 1998;43:203-9. |
| 113. | Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest 2008;118:560-70. |
| 114. | Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004;118:285-96. |
| 115. | Greten FR, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett 2004;206:193-9. |
| 116. | Bernet A, Mazelin L, Coissieux MM, Gadot N, Ackerman SL, Scoazec JY, et al. Inactivation of the UNC5C Netrin-1 receptor is associated with tumor progression in colorectal malignancies. Gastroenterology 2007;133:1840-8. |
| 117. | Paradisi A, Maisse C, Bernet A, Coissieux MM, Maccarrone M, Scoazec JY, et al. NF-kappaB regulates netrin-1 expression and affects the conditional tumor suppressive activity of the netrin-1 receptors. Gastroenterology 2008;135:1248-57. |
| 118. | Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Adv Drug Deliv Rev 2007;59:1073-83. |
| 119. | Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med 2006;203:1391-7. |
| 120. | Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene 2007;26:4833-41. |
| 121. | Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol 2004;11:1068-75. |
| 122. | Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983;301:89-92. |
| 123. | Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science 2003;300:489-92. |
| 124. | Borinstein SC, Conerly M, Dzieciatkowski S, Biswas S, Washington MK, Trobridge P, et al. Aberrant DNA methylation occurs in colon neoplasms arising in the azoxymethane colon cancer model. Mol Carcinog 2010;49:94-103. |
| 125. | Mihara M, Yoshida Y, Tsukamoto T, Inada K, Nakanishi Y, Yagi Y, et al. Methylation of multiple genes in gastric glands with intestinal metaplasia: A disorder with polyclonal origins. Am J Pathol 2006;169:1643-51. |
| 126. | Steinbach G, Lynch P, Phillips RK, Wallace MH, Hawk E, Gordon GB, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000;342:1946-52. |
| 127. | Grimm GR, Dirisina R, Lee G, Tang Y, Manjali J, Hanson NB, et al. 5-ASA suppressed induction of dysplasia on the AOM/DSS model: Correlation with inhibition of Wnt/beta-catenin transcriptional activity. Gastroenterology 2005;128:A200. |
| 128. | Suzuki S, Sakamoto S, Mitamura T, Sassa S, Kudo H, Yamashita Y. Preventive effects of sulphasalazine on colorectal carcinogenesis in mice with ulcerative colitis. In Vivo 2000;14:463-6. |
| 129. | Kohno H, Suzuki R, Yasui Y, Miyamoto S, Wakabayashi K, Tanaka T. Ursodeoxycholic acid versus sulfasalazine in colitis-related colon carcinogenesis in mice. Clin Cancer Res 2007;13:2519-25. |
| 130. | Clapper ML, Gary MA, Coudry RA, Litwin S, Chang WC, Devarajan K, et al. 5-aminosalicylic acid inhibits colitis-associated colorectal dysplasias in the mouse model of azoxymethane/dextran sulfate sodium-induced colitis. Inflamm Bowel Dis 2008;14:1341-7. |
| 131. | Stolfi C, Fina D, Caruso R, Caprioli F, Sarra M, Fantini MC, et al. Cyclooxygenase-2-dependent and -independent inhibition of proliferation of colon cancer cells by 5-aminosalicylic acid. Biochem Pharmacol 2008;75:668-76. |
| 132. | Weber CK, Liptay S, Wirth T, Adler G, Schmid RM. Suppression of NF-kappaB activity by sulfasalazine is mediated by direct inhibition of IkappaB kinases alpha and beta. Gastroenterology 2000;119:1209-18. |
| 133. | Stolfi C, Sarra M, Caruso R, Fantini MC, Fina D, Pellegrini R, et al. Inhibition of colon carcinogenesis by 2-methoxy-5-amino-N-hydroxybenzamide, a novel derivative of mesalamine. Gastroenterology 2010;138:221-30. |
| 134. | Kohno H, Suzuki R, Sugie S, Tanaka T. Suppression of colitis-related mouse colon carcinogenesis by a COX-2 inhibitor and PPAR ligands. BMC Cancer 2005;5:46. |
| 135. | Rosen ED, Spiegelman BM. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem 2001;276:37731-4. |
| 136. | Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM, et al. A new ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-gamma), GW7845, inhibits rat mammary carcinogenesis. Cancer Res 1999;59:5671-3. |
| 137. | Takahashi N, Okumura T, Motomura W, Fujimoto Y, Kawabata I, Kohgo Y. Activation of PPARgamma inhibits cell growth and induces apoptosis in human gastric cancer cells. FEBS Lett 1999;455:135-9. |
| 138. | Tanaka T, Kohno H, Yoshitani S, Takashima S, Okumura A, Murakami A, et al. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res 2001;61:2424-8. |
| 139. | Kohno H, Yoshitani S, Takashima S, Okumura A, Hosokawa M, Yamaguchi N, et al. Troglitazone, a ligands for peroxisome proliferator-activated receptor gamma, inhibits chemically-induced aberrant crypt foci in rats. Jpn J Cancer Res 2001;92:396-403. |
| 140. | Su W, Necela BM, Fujiwara K, Kurakata S, Murray NR, Fields AP, et al. The high affinity peroxisome proliferator-activated receptor-gamma agonist RS5444 inhibits both initiation and progression of colon tumors in azoxymethane-treated miceInt. J Cancer 2008;123:991-7. |
| 141. | Osawa E, Nakajima A, Wada K, Ishimine S, Fujisawa N, Kawamori T, et al. Peroxisome proliferator-activated receptor gamma ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology 2003;124:361-7. |
| 142. | Su W, Bush CR, Necela BM, Calcagno SR, Murray NR, Fields AP, et al. Differential expression, distribution, and function of PPAR-gamma in the proximal and distal colon. Physiol Genomics 2007;30:342-53. |
| 143. | Chen L, Bush CR, Necela BM, Su W, Yanagisawa M, Anastasiadis PZ, et al. RS5444, a novel PPARgamma agonist, regulates aspects of the differentiated phenotype in nontransformed intestinal epithelial cells. Mol Cell Endocrinol 2006;251:17-32. |
| 144. | Curini M, Epifano F, Maltese F, Marcotullio MC, Tubaro A, Altinier G, et al. Synthesis and anti-inflammatory activity of natural and semisynthetic geranyloxycoumarins. Bioorg Med Chem Lett 2004;14:2241-3. |
| 145. | Murakami A, Nakamura Y, Tanaka T, Kawabata K, Takahashi D, Koshimizu K, et al. Suppression by citrus auraptene of phorbol ester-and endotoxin-induced inflammatory responses: role of attenuation of leukocyte activation. Carcinogenesis 2000;21:1843-50. |
| 146. | Tsai IL, Lin WY, Teng CM, Ishikawa T, Doong SL, Huang MW, et al. Coumarins and antiplatelet constituents from the root bark of Zanthoxylum schinifolium. Planta Med 2000;66:618-23. |
| 147. | Tanaka T, de Azevedo MB, Durán N, Alderete JB, Epifano F, Genovese S, et al. Colorectal cancer chemoprevention by 2 beta-cyclodextrin inclusion compounds of auraptene and 4′-geranyloxyferulic acid. Int J Cancer 2010;126:830-40. |
| 148. | Murakami A, Takahashi D, Kinoshita T, Koshimizu K, Kim HW, Yoshihiro A, et al. Zerumbone, a Southeast Asian ginger sesquiterpene, markedly suppresses free radical generation, proinflammatory protein production, and cancer cell proliferation accompanied by apoptosis: the alpha,beta-unsaturated carbonyl group is a prerequisite. Carcinogenesis 2002;23:795-802. |
| 149. | Murakami A, Hayashi R, Tanaka T, Kwon KH, Ohigashi H, Safitri R. Suppression of dextran sodium sulfate-induced colitis in mice by zerumbone, a subtropical ginger sesquiterpene, and nimesulide: separately and in combination. Biochem Pharmacol 2003;66:1253-61. |
| 150. | Kim M, Miyamoto S, Yasui Y, Oyama T, Murakami A, Tanaka T. Zerumbone, a tropical ginger sesquiterpene, inhibits colon and lung carcinogenesis in mice. Int J Cancer 2009;124:264-71. |
| 151. | Ravichandran K, Velmurugan B, Gu M, Singh RP, Agarwal R. Inhibitory effect of silibinin against azoxymethane-induced colon tumorigenesis in A/J Mice. Clin Cancer Res 2010;16:4595-606. |
Authors
Prof. Sergio Morini, MD, Professor of Human Anatomy. Head of the Laboratory of Microscopic and Ultrastructural Anatomy, Centre for Integrated biomedical Research – CIR; University Campus Bio-Medico of Rome (Italy).
Dr. Emanuela Massi, PhD, Postdoc Fellowship for scientific collaboration at University Campus Bio-Medico of Rome, School of Medicine. Dr. Massi received PhD in Experimental Oncology, University of Foggia (Italy), School of Medicine. She studied the optimization of plasmids and protocols for DNA vaccination against murine indolent B-cell lymphoma. She also received a EACR Travel Fellowship Award for mutational analysis of EXT genes by DHPLC, at Prof W Wuyts’ lab, Department of Medical Genetics, University of Antwerp, Belgio. Recently her studies are focused on molecular pathology with main interest on genetic and epigenetic mechanisms of carcinogenesis.
Dr. Maria Luana Poeta, Assistant Professor. She received the MD degree in 2002 (University Campus Bio-Medico of Rome, Italy). In 2003 she started a Research Fellowship in the Head and Neck Cancer Research Laboratory at the Johns Hopkins University, Baltimore MD, USA, directed by Prof. David Sidransky. During a period of almost four years at the JHU she focused on studying TP53 mutations in primary tumors and surgical margins to elucidate how the different TP53 mutations impact the survival of Head and Neck cancer patients. In 2009 she completed the Residency in Clinical Pathology (Universitΰ Cattolica del Sacro Cuore, Rome, Italy) and in the same year she held the position of Assistant Professor in the University of Bari, Department of General and Environmental Physiology.
Dr. Emanuela Signori, Phd, is currently a Researcher at the National Research Council (CNR), Institute of Translational Pharmacology (IFT), and she is an Acting Professor of General Pathology at the University Campus Bio-Medico of Rome, School of Medicine. She received her PhD in Experimental Oncology at University of Foggia (Italy), School of Medicine. She actually leads the Laboratory of Molecular Pathology and Experimental Oncology at CNR. Her major research interest is the identification of tumour antigens and expression studies of genes and proteins related to tumour and/or to inflammatory diseases, in order to clarify their role in cancer progression and invasiveness and to develope DNA vaccines delivered by Electrotransfer in animal models of human diseases.
Dr. Simone Carotti, MD, PhD in Experimental and Clinical Hepatology. Laboratory of Microscopic and Ultrastructural Anatomy, Centre for Integrated biomedical Research – CIR; University Campus Bio-Medico of Rome (Italy).
Dr. Loredana Cecchetelli, Director R&D, Istituto Biochimico Italiano Giovanni Lorenzini SpA.
Dr. Mariangela De Robertis, PhD. She received her master degree in Biotechnology in 2006 (University of Bari, Italy). In 2010 she obtained her PhD in Experimental Oncology from University of Foggia (Italy) and she started a one-year postdoctoral research fellowship at Dr. J. Garcia-Foncillas’ lab, CIMA (Center for Applied Medical Research), University of Navarra, Pamplona (Spain). She is currently postdoctoral fellow at University Campus Bio-Medico of Rome, Laboratory of Molecular Medicine and Biotechnology. Her main research is focused on molecular oncology with particular attention for epigenetic modifications arising during tumor development. In the last years she has been interested in the study of colorectal carcinogenesis in a mouse model of chemically-induced colorectal cancer.
Prof. Vito Michele Fazio, MD, Specialist in Oncology, is Full Professor of Laboratory Medicine, Chairman of General Pahology, Chief Laboratory of Molecular Medicine and Biotechnology, Director Residency in Clinical Pathology, University Campus Bio-Medico of Rome, School of Medicine, and Director of the Laboratory of Oncology, Scientific Institute and Hospital (IRCCS) “Casa Sollievo della Sofferenza”, Opera di San Pio da Pietrelcina, San Giovanni Rotondo (FG), Italy.
Figures
[Figure 1], [Figure 2], [Figure 3], [Figure 4]
Tables