Soley Bayraktar1, Sameer Batoo2, Scott Okuno2, Stefan Glück3
1 Department of Medicine, Division of Medical Oncology and Hematology, Mayo Clinic Health System, Eau Claire, WI, USA; Department of Medicine, Division of Medical Oncology and Hematology, Biruni University School of Medicine, Istanbul, Turkey
2 Department of Medicine, Division of Medical Oncology and Hematology, Mayo Clinic Health System, Eau Claire, WI, USA
3 Vice President Global Medical Affairs, Early Assets, Celgene Corporation, Summit, NJ, USA
| Date of Submission | 02-Mar-2019 |
| Date of Acceptance | 09-Apr-2019 |
| Date of Web Publication | 23-May-2019 |
Correspondence Address:
Soley Bayraktar
Mayo Clinic Health System, Albert J. And Judith A. Dunlap Cancer Center, 1221 Whipple St., Eau Claire, WI 54702
Source of Support: None, Conflict of Interest: None
DOI: 10.4103/jcar.JCar_2_19
Abstract
The idea of using the immune system to fight cancer is over 100 years old. A new molecular approach led to a better understanding of the immune system. Checkpoint regulation, understanding the roles of Tregs, Th1, and Th2, development of Chimeric antigen receptor (CAR)-T cells, as well as regulation of dendritic cells and macrophages, are just a few examples of our understating that has also led to the discovery of immune checkpoint inhibitors (ICIs) and modulators. This led the Nobel Prize committee in 2018, to award Dr. James P. Allison the Nobel Prize in medicine for the discovery of Cytotoxic T-lymphocyte-associated antigen-4, and Dr. Tasuku Honjo for the discovery of programmed cell death-1 (PD-1)/PD-1-ligand (PDL-1). Several ICIs are already approved by the regulatory authorities, and many more are currently used in studies of several solid tumors and hematologic malignancies. Positive studies have led to the US Food and Drug Administration (FDA) and European Medicines Agency approval of a number of these compounds, but none to date are approved in breast cancer (BC). Moreover, PD-1/PDL-1, MSI high (and dMMR), and tumor mutational burden are the currently “best” predictive markers for benefit from immunotherapy. BCs have some of these markers positive only in subsets but less frequently expressed than most other solid tumors, for example, malignant melanoma or non-small cell lung cancer. To improve the potential efficacy of ICI in BC, the addition of chemotherapy was one of the strategies. Many early and large clinical trials in all phases are underway in BC. We will discuss the role of immune system in BC editing, and the potential impact of immunotherapy in BC outcomes.
Keywords: Breast cancer, checkpoint inhibitors, cytotoxic T-lymphocyte-associated antigen-4, immunotherapy, programmed cell death-1, programmed cell death ligand-1
How to cite this article:
Bayraktar S, Batoo S, Okuno S, Glück S. Immunotherapy in breast cancer. J Carcinog 2019;18:2
How to cite this URL:
Bayraktar S, Batoo S, Okuno S, Glück S. Immunotherapy in breast cancer. J Carcinog [serial online] 2019 [cited 2021 Oct 13];18:2. Available from:
Introduction
Evading immune destruction is an emerging hallmark of cancer. The immune system plays a dual role in cancer: it not only can suppress tumor growth by destroying cancer cells or inhibiting their outgrowth but also promotes tumor progression either by selecting for cancer cells that are more fit to survive in an immunocompetent host or by establishing conditions within the tumor microenvironment that facilitate tumor outgrowth. Nonetheless, numerous studies have shown that tumors can be recognized and contained for extended periods by the immune response through the concerted action of the innate (via chronic inflammation orchestrated by the innate immune system) and adaptive immune responses.[1] Despite these efforts, often cancer still develops, at increased frequency with age, as a consequence of selecting less immunogenic tumor cells, or the increased effectiveness of tumor-mediated immunosuppression (immune subversion) or both.[2],[3]
Our understanding of the complex interplay between cancer and the immune system has improved substantially by moving from the concept of “immune surveillance”[4] to that of “immunoediting.”[3] The concept of cancer immunosurveillance was first proposed in 1909 by Ehrlich who suggested that evolving tumors are constantly identified and eradicated by the host immune system even before clinical manifestations occur. This concept was refined by Burnet in 1970 with their proposal that genetic changes leading to malignancy are common in somatic cells and that the immune system is responsible for eliminating or inactivating these potentially dangerous mutant cells.[4] This concept has now been experimentally confirmed, primarily through demonstration of the increased incidence of malignant tumors in immunodeficient mice or humans.[5] Studies have shown that severely immunocompromised mice, with deficiencies in the innate and the adaptive immune system, have a significantly increased incidence of tumors, suggesting that immunosurveillance is essential to control the gradual development of tumors.[6]
For example, recipients of solid organ transplants typically experience cancer rates similar to nontransplanted people 20–30 years older, and risk is inversely related to age, with younger recipients experiencing a far greater relative increase in risk compared with older recipients (risk increased by 15–30 times for children, but 2-fold for those transplanted >65 years).[7] Reasons for the increased risk of most cancers after transplantation are likely owing to the interplay of several factors: The organ transplanted, prior and new exposure to viral infections, total load, and duration of immunosuppression, perhaps the specific components of the immunosuppressive regimen.
However, the fact that malignant tumors also develop in patients with a fully functional immune system suggests that immunosurveillance is only a part of the process; and as a consequence, the concept of immunosurveillance has been adapted and refined over the last 15 years into a theory termed “immunoediting,”[8] a term that well describes the dual host-protecting and tumor-promoting actions of the immune system and has three phases: elimination, equilibrium, and escape. However, these are not, in fact, separate phases, but rather represent a continuum of the interplay between tumor and immune system, shifting between elimination, equilibrium, and escape depending on the state of the immune system and genuine or acquired properties of the tumor cells.
The elimination phase in the immunoediting hypothesis represents a modern view of immunosurveillance. In the equilibrium phase, although the immune system has failed in eliminating all clinically detected tumors, it can be actively and effectively engaged to keep the tumor cells in a dormant state for many years and therefore reduce the risk of metastatic spread.[9] Finally, tumor cells may escape from immune control and proliferate in an unrestricted manner, leading to clinically apparent tumors. This escape can be mediated through various mechanisms, such as reduced immune recognition, increased resistance to attack by immune cells or the development of an immunosuppressive tumor microenvironment.[2] An example of tumor immunoediting in triple-negative breast cancer (TNBC) is provided by the presence of CASP8 mutations,[10] which can abrogate the death induced by cytotoxic CD8+ T cells, and has been described as a common mechanism of immune escape in many solid tumors.[11]
There is also increasing evidence that tumors are able to create an immunosuppressive microenvironment and recruit specific immune cells that favor tumor growth and progression. Elevated levels of CD4+ regulatory T cells (Tregs) are often found in human tumors and are associated with poor prognosis. For example, Forkhead box P3 (FOXP3)+ Treg cells are crucial for the induction and maintenance of tolerance to self-antigens. While exerting their function, Treg cells can also suppress immune responses to tumor antigens.[12] Supporting this concept, in humans, FOXP3 expression in breast cancer (BC) was associated with worse distant metastases-free survival, and the risk increased with increasing FOXP3 immunostaining intensity.[13]
In a recent paper, Ahmadzadeh et al.[14] analyzed the dominant intratumoral Treg clones for antigen specificity. In Tregs from one patient’s metastatic melanoma tumor, 6 of 11 analyzed T-cell receptors (TCRs) specifically recognized the tumor, with 3 of these being among the 10 dominant FOXP3+ clonotypes. Conventional T cells from the same tumor showed 7 of 9 clones reacting to the tumor, with 3 of these being among the 10 dominant clonotypes in the intratumoral conventional T-cell subset. Furthermore, the researchers asked if tumor-specific Tregs could also be found in peripheral blood. Analyzing the TCRβ deep sequencing data from patient 3107, they found that 6 tumor-reactive Treg TCRs (including the TCR specific for the mutated ANXA1) were found in circulating Tregs. These results suggest that peripheral blood could be a source of tumor-reactive and neoantigen-specific Tregs. Overall, the results of this study demonstrate that the elevated levels of Tregs in the tumors are likely due to tumor antigen stimulation and the resulting clonal expansion. Intratumoral Tregs displayed a TCR repertoire that was distinct from conventional T cells but overlapped with circulating Tregs. Most importantly, the most dominant intratumoral Treg TCR clones showed reactivity to the tumor and patient-specific neoantigens. The identification of Treg specificity may inform the design of future TCR-based immunotherapies against cancer. In [Figure 1] are reported major functions and components of the immune system relevant for potential BC therapy.
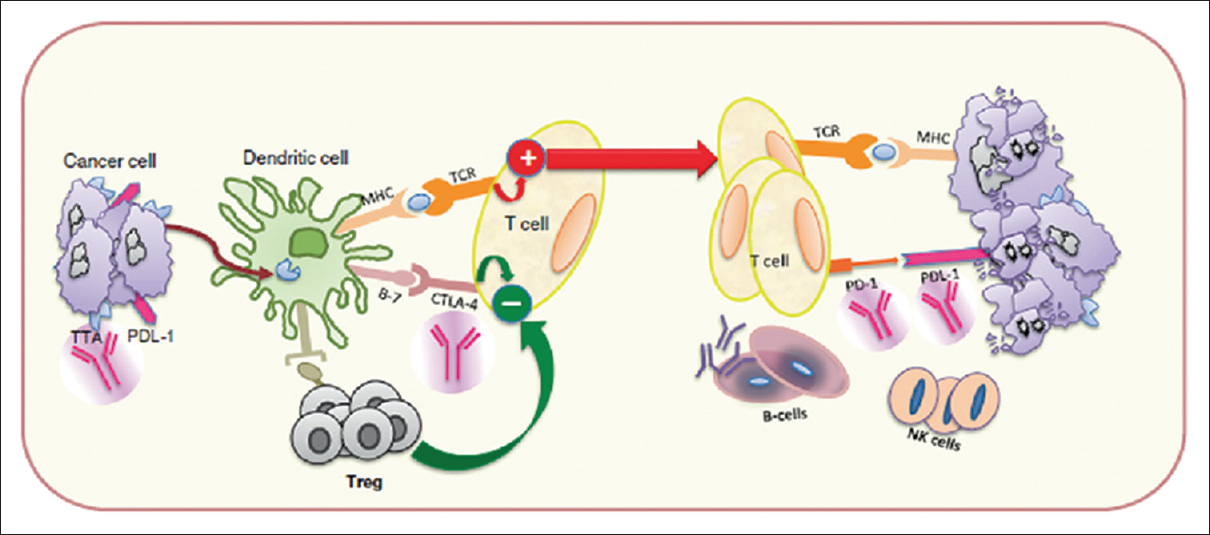 |
Figure 1: Immune system functions and components relevant to breast cancer therapy[95] Click here to view |
Immune Checkpoints
T-cells are activated by foreign antigens presented on major histocompatibility complex and the co-expression of TCR on the one hand and by a concurrent coactivation of costimulatory and/or co-inhibitory signals on the other hand [Figure 2]. The latter includes members of the CD28/B7 family and is known as “immune checkpoints.”[15],[16]
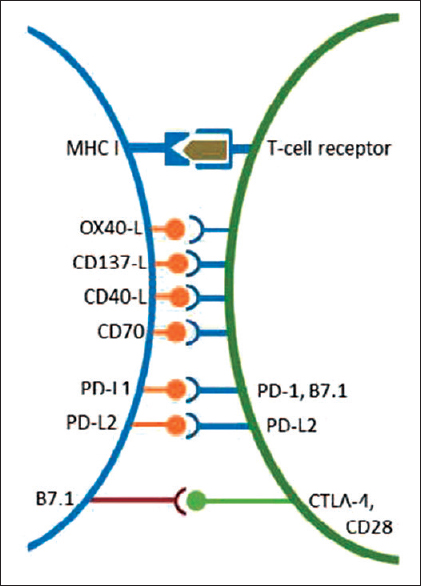 |
Figure 2: Co-stimulatory and co-inhibitory receptors expressed by T-cells (green) and target cells (rose). Reproduced with permission from Schutz F. et al. PD-1/PD-L1 pathway in Breast Cancer, Oncol Res Treat 2017 Click here to view |
Immune checkpoints are involved in T-cell tolerance as well as activation. They play a crucial role in maintaining self-tolerance and immune homeostasis under physiological conditions, thereby protecting tissues from unnecessary damage when the immune system has efficiently cleared the pathogen.[17] Even maternal immune tolerance toward the fetus is in part regulated by checkpoint inhibitors.[18]
Tumors may express immune inhibitory signals resulting in an attenuated immune reaction against the pathologic antigens.[19] Cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), the programmed cell death-1 and its ligands (PD-1/PD-L1/2) axis, lymphocyte activation gene-3, and T-cell immunoglobulin mucin-3 are negative signals inhibiting T-cell immune response. In the context of tumor immunology, CTLA-4 signaling is more involved in limiting the initiation of a T-cell response in the lymph nodes, while PD-1 features more prominently later on in the process and serves to limit T-cell activity in the tumor microenvironment.[20] After TCR engagement, CTLA-4 is upregulated to attenuate T-cell responses and prevent expansion of autoreactive T cells, primarily during the priming phase within the lymph nodes. Anti-CTLA antibodies such as ipilimumab and tremelimumab were both tested in solid tumors including BC with limited efficacy.[21],[22]
Programmed Cell Death-1/programmed Cell Death Ligand-1 Pathway
PD-1 is an inhibitory immune checkpoint inhibitor (ICI) which is expressed on the surface of T cells, B cells, natural killer (NK) T cells, T-cell lysis, and causes induction of tolerance to antigens.[23],[24],[25]In vitro blockade of PD-1 with monoclonal antibodies (mAbs) led to a 2-fold increase in cytokine production.[26] However, thein vivo activity also depends on T-cell motility as well as the duration of the interaction with antigen-presenting cells and target cells.[27] When T cells have been activated by their TCR, PD-1 is expressed simultaneously to offer the attacked cell a way of escaping the immune reaction. PD-1 decreases once the immune response has eliminated the pathologic antigen.[28]
In solid tumors, the PD-1/PD-L1 inhibitory pathway cause inactivation of the immune system by increasing the expression of PD-L1 on the tumor cell surface.[29] PD-L1 expression has been associated with large tumor size, high grade, high proliferation, estrogen receptor (ER)-negative status, and human epidermal growth factor receptor-2 (HER2)-positive status,[30] and it is inversely correlated with survival in ovarian[31] and BC.[32] PD-L1 is expressed in 20% of TNBCs.[33] This indicates that although antitumor immunity is elicited against many solid tumors, it is counterbalanced by immunosuppressive factors. It was shownin vivo that PD-L1 increases tumorigenesis and invasiveness and makes tumor cells less susceptible to specific CD8+ T-cells.[34] The goal of ICIs such as anti-CTLA-4 and anti-PD-1/anti-PD-L1 is to “release the brakes” and enhance T-cell activation by blocking negative pathways.
Furthermore, myeloid-derived suppressor cells (MDSC) play a leading role in immunosuppression in various cancer types. Accumulating evidence in recent years have even highlighted them as a major driver of an immunosuppressive tumor microenvironment.[35] The research shows that although ICI may prove to be effective, therapeutic resistance occurs in the majority of patients, leading to tumor progression.[36] This occurs due to the immunosuppressive tumor microenvironment represented by several immunosuppressive factors and cells, including MDSC. Importantly, the efficacy of cancer immunotherapy has been reported to be negatively correlated with an increased MDSC frequency and function.[37] Therefore, MDSC could be a promising target in cancer immunotherapy, especially in combination with ICI.
Prognostic Value of Immune-Related Gene Signatures
In past years, gene expression profiling has been used in an effort to more precisely define BC taxonomy and identify prognostic and predictive signatures.[38] The common denominator between the majority of the “first generation” signatures is their overall capacity to detect subtle differences in the cell cycle and proliferation. For this reason, they have not been found to be prognostic in the triple-negative or HER2+ subtypes since these tumors are by “nature” highly proliferative. Several investigators have tried to overcome the limitations of these “ first generation” signatures by focusing on the BC microenvironment or immune response (or both) to define promising “second generation” prognostic signatures.[39] Unsupervised gene expression profiling of cancer-associated stroma revealed a signature enriched for CD8+ T-cell responses that was predictive of good prognosis.[40] An immune response module, the STAT1 module, has been shown to be associated with survival in patients with TNBC and HER2+ BC,[41],[42] and in the same BC subtypes, the overexpression of immune-related genes was able to identify subgroups of patients with a better prognosis.[43],[44] Similarly, in other studies, the high expression of B-cell and immunoglobulin-based metagenes is associated with a low risk of developing distant metastases in patients with untreated ER-negative breast tumors,[42],[45] whereas in patients with untreated ER-negative HER2-positive BC, elevated expression of the STAT1-related and T-cell-related metagenes is associated with a low risk of distant metastasis.[46] These studies suggest that effective engagement of the immune system, although insufficient to eliminate the tumor, can help to reduce the risk of tumor spreading, or maintain tumor dormancy.[9]
The Role of the Lymphocytic Infiltrate in Breast Cancer
The presence of tumor-infiltrating lymphocytes (TILs) is observed in some breast tumors and has been reported to be a good prognostic feature for certain types of BCs,[43],[47] particularly for ER-negative tumors and HER2-positive subtypes.[48] Furthermore, TILs have been negatively correlated with the patient’s age at diagnosis.[49] More recently, the nature of TILs has been better characterized. Ruffell et al.[50] have reported that TILs are composed mainly of CD3+/CD56− T cells but that a minority consisted of NK cells or CD20+ cells. The majority of CD3+ cells were either CD4+ or CD8+ T cells. Interestingly, CD8+ cells did not express granzyme B at baseline, which means that they did present inactivation status, but they did express granzyme B after neoadjuvant chemotherapy in one-third of the patients. Finally, a minority of TILs presented T and NK-cell features.[50] The genomic characteristics of TIL+ tumors are important to understand which molecular mechanisms lead to lymphocyte infiltration. Genomic instability may promote anti-tumor immune response through tumor-associated antigens (TAAs). Some mechanisms of chemokine release by the tumor have been described and correlated with lymphocyte attraction. TILs have been associated with CXCL9 and CXCL13 expression by the tumor.[48] TIL + tumors present a specific methylation pattern on immune-related genes, including CCL5,[51] and a cluster of chemokines is lost in a subset of BC.[52]
In TNBC and HER2-positive BC, the association between the presence of TILs or expression of immune markers and the likelihood of achieving a pathologic complete response (pCR) after neoadjuvant chemotherapy is consistent and strong. A high level of expression of immune markers is associated with different immune cell types and functions and has been associated with benefit from chemotherapy in TNBCs.[30],[46],[53] This association has also been confirmed by evaluation of the TIL density.[48],[54] Overall, these data suggest that the immune system collaborates with the action of chemotherapy in TNBCs, as suggested by data from preclinical studies.[55],[56] However, whether drug-specific immunomodulation properties,[56] such as inducing immunogenic tumor cell death (postulated for anthracyclines) or engaging different immune effector mechanisms, are associated with different clinical outcomes is unknown and is currently an active area of investigation. Interestingly, assessments of the immune microenvironment after neoadjuvant chemotherapy in patients with TNBC with residual disease has shown that the immune microenvironment can be turned from “cold” (containing few TILs) to “hot” (higher TIL presence) in some patients.[57] Tumors that remain or become “cold” after chemotherapy have a higher risk of relapse compared with tumors that remain or become “hot.”[57] These data also support the concept of chemotherapeutic agents having immunomodulatory activity, and thus, acting as an immunological stimulant in the tumor microenvironment to induce antitumor immunity.[57] Whether the immune system has different prognostic and predictive roles in different molecular subtypes of TNBCs has not been defined yet.
Overall, considering that, in TNBCs, a “hot” immune microenvironment is associated with a better prognosis and a higher likelihood of benefit from chemotherapy, it should not be surprising that many investigators have identified a strong association between high levels of immune markers or TILs and a low risk of relapse and/or death in patients with early-stage TNBC treated with systemic chemotherapy.[58] These results suggest that, in TNBC, the risk of recurrence in the early disease setting can be effectively defined by adopting the appropriate immune markers for risk stratification. These results also distinguish a subgroup of patients with TNBC characterized by a “cold” immune microenvironment that has a high risk of relapse, despite treatment, and a low likelihood of benefit from cytotoxic chemotherapy.[46] Evidence for the clinical utility of TIL evaluation, however, is still scarce, in part because TIL assessment lacks sufficient standardization; however, efforts to improve consistency and reproducibility are underway.[59]
Immunogenicity of Breast Cancer
BC has not been traditionally considered immunogenic, as opposed to melanoma and renal cell carcinoma, which have been traditionally considered more responsive to immunotherapies. Moreover, the tumor microenvironment in BCs releases immune-suppressive factors that make the antigen presentation difficult and that have a negative impact on the immune response.[60] Furthermore, by blocking endogenous immune checkpoints that normally terminate immune responses after antigen activation, it is possible to evade immune destruction.
However, it seems that, despite a weak influence on primary tumor growth, the immune system may be effective in preventing BC metastases.[61],[62] It seems that any tumor could be immunogenic with appropriate immune activation. In a neoadjuvant clinical trial (Trial of Principle study) in which patients with ER-negative BCs were treated with anthracycline monotherapy, high immune module scores were associated with sensitivity to anthracyclines.[53] The immune system seems also to be pivotal in determining the response to mAbs and tyrosine kinase inhibitors, and some evidence indicates a possible role in the response to endocrine treatment. Antibody-dependent cellular cytotoxicity has long been implicated as one of the mechanisms of action for trastuzumab.[63],[64] Therefore, complete tumor response after molecular targeted therapies requires a functioning immune system, pointing the way toward radically new combination therapies with a targeted and immune approach.[65] The mAbs against antigen tumor target or immune-regulatory molecules, cell-based therapies including adoptive transfer of ex vivo-activated T cells and NK cells, or blockade of Treg cells could be useful to amplify the anti-tumor response.
Tumor Mutational Burden and Mutational Signatures in Breast Cancer
The use of immunotherapy is exponentially increasing in the treatment of patients with advanced solid tumors. However, the response rates vary significantly between different tumor types and even within the same tumor type (e.g., in lung cancer approximately 1 in 4 patients respond to immunotherapy). In order to better identify patients that will respond to immunotherapy, several markers have been proposed. Tumor mutational burden (TMB) has emerged more recently as a quantitative marker that can help predict responses to immunotherapies across different cancers, including melanoma, lung cancer, and bladder cancer and BC.[66] TMB is a measure of the overall number of somatic protein-coding mutations occurring in the tumor specimen. Bonta et al.[67] analyzed 54 patients with solid tumors treated with immunotherapy for which they had genomic sequencing (FoundationOne). There were 39 lung cases and 15 nonlung (gastrointestinal, genitourinary, sarcoma, and breast). Among patients with known TMB, 60% (18/30) had a favorable response (stable disease or response to therapy). Higher TMB values were correlated with increased probability of a favorable response. In their study, a TMB cutoff value of 8 mutations (mut)/megabase (MB) yielded a sensitivity of 95% and a specificity of 58% for predicting favorable response. In 2018, ASCO annual meeting, Barroso-Sousa et al.[68] evaluated mutational load across BCs. Samples were classified as having high TMB if they had >10 mut/MB. They included 3689 samples for the analysis. The median TMB was 1.55 mut/MB. TMB significantly varied according to histology (ductal > lobular, P = 4.6 × 10–13), tumor subtype (HR-/HER2+ > TNBC > HR+/HER2+ > HR+/HER2-, P < 0.05), staging (metastatic > primary, P = 2.2 × 10–16) and site of metastasis (higher soft tissue, and lowest lung, P < 0.05). They found a total of 70 (~2%) hypermutated tumors (62.8% metastatic vs. 37.2% primary samples). Mutational signature analysis of the hypermutated samples showed the presence of dominant APOBEC (77.1%), homologous recombination (HR; 2.9%), defective DNA mismatch repair (MMR; 18.6%), and POLE hypermutation (1.4%) signatures. Median TMB was higher for samples with POLE and HR signature, followed by those with MMR and APOBEC (93.1, 38.7, 14.6, and 12.4 mut/MB, respectively). Among hypermutated tumors, eight samples had somatic mutation in the POLE gene but only the case with POLE signature high had a characterized POLE driver mutation. In addition, 80% of hypermutated tumors with APOBEC signature had PIK3CA mutations versus 31% of hypermutated tumors with other signatures (P = 0.0005). In another study, Xu et al.[69] aimed to predict the level of TMB in patients with BC by the expression of ER, PR, HER-2, and Ki-67, thereby anticipating the prognosis of patients and the possible response to immunotherapy. HER-2 expression positivity was significantly associated with TMB (HER-2 positive vs. HER-2 negative, odds ratio [OR] = 34.81, 95% confidence interval [CI]: 3.711–821.689, P = 0.0065). Furthermore, higher TMB was distributed in the patients who were Ki-67 expression positive (>14%) than those who were Ki-67 expression negative (≤14%) (OR = 0.217, 95% CI: 0.054–0.806, P = 0.0242). However, no significant differences of TMB were found between ER-positive group and ER-negative group (OR = 3.133, 95% CI: 0.124–127.687, P = 0.4954) and between PR-positive group and PR-negative group (OR = 1.702, 95% CI: 0.162–20.335, P = 0.6492). In a multivariate analysis, high TMB (>5.56) was an independent predictive factor for decreased disease free survival (DFS) (adjusted hazard ratio [HR], 5.594; 95% CI: 1.694–18.473; P = 0.005). These results suggest that the level of TMB value can be predicted based on the expression levels of ER, PR, HER-2, and Ki-67, which may indicate the prognostic and predictive value of immunotherapy in patients with BC.
Immunotherapy for Triple-Negative and Human Epidermal Growth Factor Receptor 2-Positive Breast Cancers
Immunotherapy with checkpoint inhibitors has made a significant impact in the treatment of melanoma, renal cell carcinoma, and non-small cell lung cancer (NSCLC) in recent years.[70],[71],[72],[73] New agents such as nivolumab and pembrolizumab (a fully human IgG4 programmed death 1 [PD-1] ICI antibody) selectively blocks the interaction of the PD-1 receptor with its two known programmed death ligands, PD-L1, and PD-L2, disrupting the negative signal that regulates T-cell activation and proliferation.[74] Anti-PD-L1, such as atezolizumab is the first FDA approved PD-L1 blocker and has been used as first-line treatment for cisplatin-resistant metastatic urothelial carcinoma, metastatic NSCLC. The variety of clinical trials is under progress for colorectal cancer, bladder cancer, renal cell carcinoma, head and neck cancer, and GI malignancies. Durvalumab (MEDI4736), another PD-L1 blocker, binds to PD-1 and CD80, is approved in unresectable NCSLC and locally advanced or metastatic urothelial carcinoma. Avelumab is also another PD-L1 blocking antibody indicated for the treatment of adults and pediatric patients 12 years and older with metastatic Merkel cell carcinoma.
As mentioned before, there is preliminary evidence of positive correlation between high mutational burden of tumors and clinical benefit from immunotherapy strategies (i.e., checkpoint inhibitors anti-CTLA-4 and anti-PD-1 antibodies), with remarkable effects seen with tumors displaying the highest rates of mutations such as melanoma.[75],[76] This is also illustrated by the antitumoral immunologic response to anti-PD-1 antibody in patients with colorectal cancer and increased mutational burden secondary to mismatch repair deficiency.[77]
As discussed above, in recent years, the better knowledge of BC biology has provided an opportunity to develop some types of immunotherapy to overcome the relative nonimmunogenic property of BC and improve immune response. TNBCs have a higher number of TILs[78] and higher PD-L1 protein[79],[80] or mRNA[33],[58] expression compared with other BC subtypes. PD-L1 expression is significantly associated with the presence of TILs,[80] which suggests that the most common mechanism of regulation of PD-L1 expression in TNBC is regulatory feedback (acquired resistance) to immune engagement. An extremely heterogeneous pattern of immune infiltration, however, has been described among TNBC subtypes.[81] A significant association has also been found between PD-L1 mRNA expression and the presence of PD-L1 copy-number alterations, with basal-like BC having the highest frequency of PD-L1 gains/amplifications (17%).[30] In addition, the loss of PTEN expression in TNBCs is associated with PD-L1 overexpression,[33] confirming an association between increased PI3K signaling and the presence of PD-L1.
The finding that a population of TNBC is immunogenic and actively engaged by the immune system provides a strong rationale for testing immunotherapies in this type of BC. The potential importance of immune checkpoint guided therapy in TNBC is underscored by recent reports. Several Phase I trials with immune-checkpoint inhibitors in patients with advanced-stage TNBC have been reported.[82] In one of them, 188 patients with advanced-stage TNBC positive for PD-L1 expression were treated with the anti-PD-1 mAb pembrolizumab with an overall response rate (ORR) of 18.5% (five out of 27 patients). Seven additional patients had stable-disease. Of the screened patients, ≥1% PD-L1 expression was detected using IHC labeling of stromal or tumor cells in archival specimens from 58% of patients with the 22C3 antibody. Avelumab, an anti-PD-L1 IgG1 antibody, showed modest anti-tumor activity among 57 patients with TNBC with only 5 partial responses (PR) observed (8.8%; 95% CI: 2.9, 19.3).[83] In patients with TNBC who had PDL1+ immune cells within the tumor, 44.4% (4 of 9) had PR, compared with 2.6% (1 of 39) PD-L1-negative immune cells. In another Phase I study,[84] selectively enrolled cohort of patients with PD-L1-positive metastatic TNBC patients received MPDL3280A, a human anti-PD-L1 mAb, 15 or 20 mg/kg IV every 3 weeks for up to 1 year. The ORR was 33%, including 1 complete response and 2 PRs. Responders included patients with visceral metastases at baseline. At the time of the clinical data cutoff, all responses occurred within 6 weeks of the first dosing of MPDL3280A, and all of the responses were ongoing. The median duration of response had not been reached (range: 18 to 56+ weeks). Different trials are ongoing to establish the safety and clinical activity of ICIs in both PD-L1-positive and PD-L1-negative TNBCs, alone or in combination with chemotherapies [Table 1].
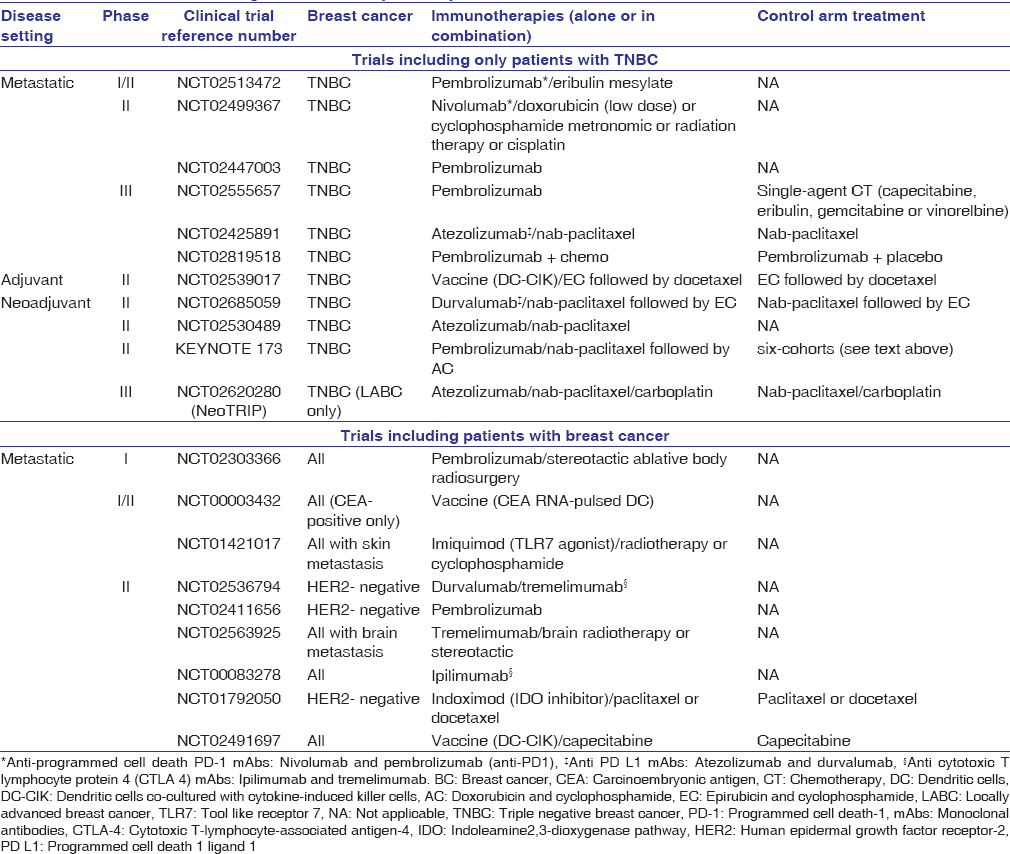 |
Table 1: Clinical trials testing immunotherapies in patients with breast cancer Click here to view |
In light of the promising preliminary results obtained with immune-checkpoint inhibitors, their expected curative potential[85] and their beneficial safety profile, these agents are already being assessed for the treatment of patients with early-stage TNBC. Three trials in patients with Stage I–III TNBC are currently ongoing to evaluate the potential activity of immune-checkpoint inhibitors in combination with chemotherapy in the neoadjuvant setting. In the Phase III trial NeoTRIPaPDL1 (NCT02620280), patients with locally advanced TNBC will be randomly assigned to receive nab-paclitaxel and carboplatin with or without a PD-L1-inhibitor (atezolizumab). Notably, the primary endpoint will be event-free survival. A Phase II trial will evaluate atezolizumab in combination with nab-paclitaxel (NCT02530489). Finally, a Phase I/II trial will test the safety and efficacy of durvalumab, another anti-PD-L1 antibody, in combination with weekly nab-paclitaxel followed by dose-dense chemotherapy containing cyclophosphamide and doxorubicin (NCT02489448).
Another interesting immune molecule is CTLA-4 (CD152), which is similar to PD-1, but its immune inhibitory signals are different. CTLA-4 knockout mice display early lethality, unlike PD-1 knockouts, which demonstrate late-onset and organ-specific autoimmunity. Anti-CTLA4 mAb treatment has shown robust tumor responses in Phase III trials, but with considerable adverse events.[86] Still, combining anti-CTLA-4 mAb with trastuzumab has shown synergy in preclinical mouse models.[87] Hence, immune-therapeutics that augment CD8 T-cell anti-tumor activity – such as anti-PD1 and anti-CTLA4 mAbs – given in combination with trastuzumab in patients with HER2+ BC may improve outcome by enhancing host immunity.[65],[88],[89] Given this evidence, the evaluation of baseline immune response and the identification of surrogate markers of immune system activation could be helpful in the management of BC to identify patients who may benefit from these combination therapies, even eliminating the need for combination cytotoxic chemotherapy.
It is important to note that a subset of patients treated with ICIs experience an accelerated tumor growth rate compared with pretreatment kinetics, known as hyperprogression. Kanjanapan et al.[90] explored the relationship between hyperprogressive disease (HPD), treatment-related toxicity, and clinical factors. They observed a 7% rate of HPD within a range of solid tumors treated with ICIs, comparable to other reports. There was no association between HPD and clinically significant adverse events, age, tumor type, or type of therapy. We need further studies to identify predictors of HPD.
Immunotherapy for Estrogen Receptor-Positive Breast Cancer
The immunotherapy has so far shown more limited responses in ER-positive BCs compared to TNBCs that tends to overexpress PDL1 as outlined above. ER + BCs may be less immunogenic, with inadequate activation of immune effector cells and/or other adaptations in the tumor microenvironments that suppress antigenicity and/or suppress activation of the adaptive or innate immune systems. Studies to determine how to reestablish immune-based host elimination of tumors offer potential, particularly for the eradication of the many small foci of growth arrested but surviving cells that remain during treatment with endocrine therapies and are the source of distant recurrences in ER + BCs.
Immunotherapy for Inflammatory Breast Cancer
The role of immune infiltrate and immune checkpoints was also investigated in relation with genomic abnormalities in IBC samples.[91] The pathological examination of 20 IBC tissue samples identified a subset of IBC tumors associated with infiltration of immune cells. IHC staining identified the majority of infiltrating cell populations as CD8+ cytotoxic T cells, and high levels of CD8+ infiltration was observed in 5/12 tumors. In order to explore the possible role of PD-L1 in IBC, the investigators performed IHC staining of IBC tissues. The evaluation of PD-L1 staining demonstrated low-intensity tumor cell staining in 3/12 tumors studied and high-intensity tumor cell staining in 1/12 tumors. PD-L1 mRNA expression has been reported to as high as 38% among patients with IBC, which is higher than non-IBC (28%) and correlates positively with pCR.[92] Notably, somatic mutation rates were significantly higher in high infiltration versus low infiltration tumors (P < 0.05).[91] The authors speculated that this correlation between somatic mutation rate and immune cell infiltration might be related to the exposure of tumor neo-antigens to the immune system. A Phase II clinical trial for patients with metastatic IBC assessing the efficacy of anti-PD-1 inhibitor monoclonal antibody (pembrolizumab) is under development (NCT02411656).
Maintenance Immunotherapy in Patients With Metastatic Breast Cancer Who Have a Clinical Benefit With Chemotherapy
Recchia Fet al.[93] investigated the effect of maintenance immunotherapy with the aim of prolonging progression-free survival (PFS) and overall survival (OS), through immune-mediated mechanisms, patients with hormone-resistant MBC who had not progressed with chemotherapy. From 1996 to 2009, 74 patients with MBC were entered into the study and received the following maintenance immunotherapy regimen: Interleukin-2 (IL-2), (1.8 M UI), and oral retinoic acid (0.5 mg/kg), 5 days/week, 3 weeks/month for 1 year. Therapy was continued, with intermittent schedule until progression. The median age was 55 years (range 31–75); 30% of patients were premenopausal; 60% were ER + and had progressed after 2 or more lines of hormonal therapy. The 74 patients had received 368 courses of chemotherapy regimens (median of 6 courses each patient). Thirty-six patients had received, also, high-dose chemotherapy with peripheral blood progenitor cell transplantation. After a median follow-up of 100 months (range 96–200), each patient had received a median of 8 courses of immunotherapy (a total of 924 courses delivered). No WHO Grade 3 or 4 toxicity was observed; Grade 2 cutaneous toxicity and autoimmune reactions occurred in 19% and 16% of patients, respectively. Statistically significant improvements were observed in the number of lymphocytes (P < 0.001), natural-killer cell count (P < 0.001), and the CD4+/CD8+ ratio (P < 0.01). A 10-year PFS and OS were 25% and 31%, respectively. Although these data needs to be matured in BC field, this approach has already been proven significant in improving PFS and OS in Stage III NSCLC patients. In February 2018, FDA approved durvalumab as a maintenance therapy for patients with unresectable Stage III non-small cell lung cancer whose disease has not progressed following concurrent platinum-based chemotherapy and radiation therapy.[94]
Vaccine-Based Therapies for Breast Cancer
Vaccines constitute an active and specific immunotherapy designed to stimulate the intrinsic anti-tumor immune response by presenting TAAs expressed on normal tissues that are overexpressed on tumor cells. Malignant cells can express both normal self-antigens and specific TAAs that arise from genetic mutations or epigenetic changes or both, recognized by the immune response through either their loss or de novo aberrant expression. Many TAAs (including MUC1, HER-2, CEA, hTER, and WT1) have been identified and been shown to be specifically recognized by T cells.[95] The induction of strong immunity by cancer vaccines is expected to lead to the establishment of immunological memory, thereby preventing tumor recurrence.
CAR T-cell therapy is an innovative form of immunotherapy wherein autologous T cells are genetically modified to express chimeric receptors encoding an antigen-specific single-chain variable fragment and various costimulatory molecules [Figure 3]. CARs are recombinant receptors that typically target native cell surface antigens.[96] Unlike the physiological TCR, which engages human leukocyte antigen (HLA)-peptide complexes, CARs engage molecules do not require peptide processing or HLA expression to be recognized. Therefore, on administration, these modified T cells traffic to, and recognize, cancer cells in an HLA-independent manner.[96]
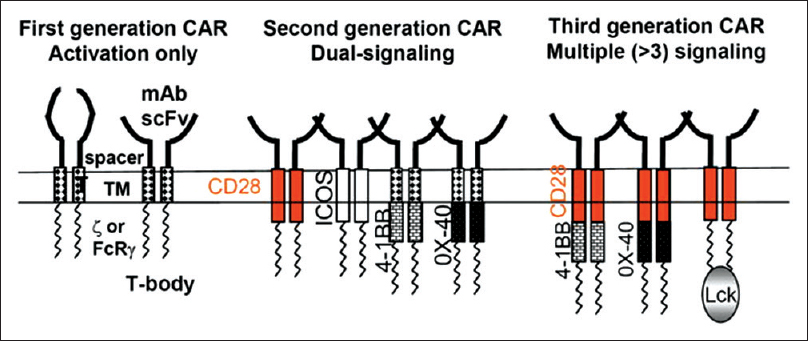 |
Figure 3: The general architecture of a chimeric antigen receptor consists of a single-chain variable fragment derived against a predetermined tumor-associated antigen followed by a CD3ζ domain required for provision of signal 1 and T-cell activation upon antigen recognitionBayraktar Click here to view |
HER2 and mesothelin are two TAAs currently under investigation. Amplification of HER2 oncogene leads to uncontrolled cell proliferation and occurs in approximately 20% of BCs.[97] Globerson-Levin et al.[98] generated a HER2-specific, second-generation CAR containing CD28, and fragment crystallizable receptor signaling domains and tested its efficacy in a syngeneic mouse mammary tumor model. Transduced T cells exhibited potent cytotoxic capacity and cytokine secretion on antigen recognition. In addition, repeated injections of HER2-directed CAR T cells eliminated spontaneous HER2-positive tumors and enhanced survival in transgenic mice. Mesothelin is a glycoprotein expressed on a broad range of solid tumors, with limited expression on normal tissues.[99] Mesothelin expression has been shown to be enriched in TNBC and is associated with poor outcomes.[100] Patients with TNBC are not suitable for targeted therapy or hormone therapy, so adoptive transfer of mesothelin-specific CAR T cells offers an alternative option. Tchou et al.[101] engineered mesothelin-specific CAR T cells and reported a cytolytic capacity against primary breast tumor cells in vitro. However,in vivo antitumor activity was not evaluated in this study.
A major therapeutic challenge to therapy in BC is acquired resistance that results from antigen escape. For instance, under selective pressure, HER2 can undergo proteolysis to cleave the extracellular domain without compromising kinase activity. One approach to overcome this limitation is to use a dual-targeting CAR system, in which engineered T cells coexpress two CARs that recognize two distinct antigens. Redirected T cells can be activated in the presence of either antigen to mitigate antigen-loss escape.[102] Alternatively, CAR T cells can be modified to secrete inflammatory cytokines, such as IL-12, or costimulatory ligands, such as 4-1BB ligand, to stimulate an endogenous immune response against tumor cells via epitope spreading.[103]
The Promise of Immunotherapy in Combination With Chemotherapy in Breast Cancer
Immunotherapies and cancer vaccines are more effective when given in combination with standard cancer treatments, which appear to increase their effectiveness by initiating tumor-specific immunity by increasing antigen presentation, MHC upregulation, increase T cell function, increased release of pro-inflammatory cytokines and cross-presentation through dendritic cells.[104],[105] The elimination of Treg cells and reduction of MDSC potentially provides another basis for the synergistic effect between cancer vaccines and chemotherapy.[105] Existing clinical data regarding chemoimmunotherapy from patients with other solid tumors have shown that patients who receive atezolizumab plus chemotherapy[106] or pembrolizumab plus chemotherapy[107] have improved OS and DFS compared to patients who receive standard chemotherapy alone.
Nab-paclitaxel as partner with ICI is particularly appealing to researchers trying to improve the chemoimmunotherapy trial results for several reasons. First and foremost, steroids which may downregulate antigen presentation are not a required premedication before nab-paclitaxel infusion;[108] it has a well-characterized and acceptable adverse event profile and has a distinct pharmacokinetics profile with higher accumulation in tumor microenvironment versus paclitaxel.
Recently, the primary results from the Phase III IMpassion130 trial[109] establish the benefit of adding ICI to nab-paclitaxel in patients with TNBC. In this Phase III trial, patients with untreated metastatic TNBC were randomly assigned to receive atezolizumab plus nab-paclitaxel or placebo plus nab-paclitaxel. In the intention-to-treat analysis, the median PFS was 7.2 months with atezolizumab plus nab-paclitaxel, as compared with 5.5 months with placebo plus nab-paclitaxel (HR: 0.80; 95% CI, 0.69–0.92; P = 0.002); among patients with PD-L1–positive tumors, the median PFS was 7.5 and 5.0 months, respectively (HR: 0.62; 95% CI, 0.49–0.78; P < 0.001). In the intention-to-treat analysis, the median OS was 21.3 months with atezolizumab plus nab-paclitaxel and 17.6 months with placebo plus nab-paclitaxel (HR: 0.84; 95% CI, 0.69–1.02; P = 0.08); among patients with PD-L1-positive tumors, the median OS was 25.0 and 15.5 months, respectively (HR: 0.62; 95% CI, 0.45–0.86). No new adverse effects were identified.
Another Phase II trial[110] results also showed the safety and improved the efficacy of concurrent administration of durvalumab (10 mg/kg) with weekly nab-paclitaxel (100 mg/m2) ×12 followed by dose-dense AC (doxorubicin and cyclophosphamide) ×4 as neoadjuvant therapy for Stage I-III TNBC. The primary efficacy endpoint was pCR. Seven of the 12 patients achieved pCR. Eight patients (25%) experienced Grade 3 adverse events including 3 patients with neutropenia (1 neutropenic fever), and one patient each with fatigue, dyspnea, line infection, transaminitis, hypertension/skin rash. No perioperative adverse events were seen.
The addition of pembrolizumab to neoadjuvant chemotherapy demonstrated promising antitumor activity and exhibited a manageable toxicity profile in a cohort of patients with early-stage TNBC, according to results of the KEYNOTE-173 trial[111] presented at 2018 San Antonio BC Symposium. All patients received a single dose of pembrolizumab 200 mg on day 1 of the first cycle. In the second to fifth cycles, patients received pembrolizumab 200 mg plus one of the six chemotherapy regimens: Nab-paclitaxel 125 mg/m2 once weekly (cohort A); nab-paclitaxel 100 mg/m2 once weekly plus carboplatin area under the curve 6 every 3 weeks (cohort B); nab-paclitaxel 125 mg/m2 once weekly plus carboplatin area under the curve 5 every 3 weeks (cohort C); nab-paclitaxel 125 mg/m2 once weekly plus carboplatin area under the curve 2 once weekly (cohort D); paclitaxel 80 mg/m2 once weekly plus carboplatin area under the curve 5 every 3 weeks (cohort E); and paclitaxel 80 mg/m2 once weekly plus carboplatin area under the curve 2 once weekly. In the sixth to ninth cycles, all patients received doxorubicin 60 mg/m2 every 3 weeks and cyclophosphamide 600 mg/m2 every 3 weeks plus pembrolizumab 200 mg every 3 weeks. Researchers reported ORRs of 100% (90% CI, 74–100) in cohorts B and C, 90% (90% CI, 61–100) in cohorts D and F, 80% (90% CI, 49–96) in cohort A, and 70% (90% CI, 39–91) in group E. Eighteen patients (30%) experienced immune-related adverse events, the most common of which were Grade 2 hypothyroidism (n = 4), Grade 1 hyperthyroidism (n = 3), Grade 3 colitis (n = 2), and Grade 3 rash (n = 2). Sixty percent (90% CI, 30–85) of all patients achieved pCR. The findings support the ongoing Phase III KEYNOTE-522 trial, which is comparing the combination with placebo in this patient population. Importantly, these ongoing trials will answer the question whether the correlation between pCR with chemo-immunotherapy and long-term outcomes is as clear as the correlation between pCR and chemotherapy in this patient population.
Conclusion
Immunomodulation seems to be a promising strategy in solid tumors. High immunogenicity has been described in BC subtypes with a high proliferation index (TNBC and HER2). Immune checkpoints are one of the major mechanisms of immune escape. Expression of PD-L1 on tumor cells leads to lower activity of CD8+ T-cells. Antibodies against PD-1 or PD-L1 are being investigated in clinical trials. First results are promising but only a subset of patients (20%) respond to immune checkpoint inhibitory treatment. Predictive markers are urgently needed to select those patients with the best chance for an effective treatment.[112] One possible avenue is immuno-molecular therapy, which integrates immune and molecular features to devise novel combinatorial approaches based on targeting intracellular molecular alterations and modulating the immune response.[113]
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
| 1. |
Disis ML. Immune regulation of cancer. J Clin Oncol 2010;28:4531-8.
 |
| 2. |
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011;331:1565-70.
|
| 3. |
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol 2002;3:991-8.
|
| 4. |
Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res 1970;13:1-27.
|
| 5. |
Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004;21:137-48.
|
| 6. |
Mapara MY, Sykes M. Tolerance and cancer: Mechanisms of tumor evasion and strategies for breaking tolerance. J Clin Oncol 2004;22:1136-51.
|
| 7. |
Webster AC, Wong G, Craig JC, Chapman JR. Managing cancer risk and decision making after kidney transplantation. Am J Transplant 2008;8:2185-91.
|
| 8. |
Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases – Elimination, equilibrium and escape. Curr Opin Immunol 2014;27:16-25.
|
| 9. |
Pusztai L, Karn T, Safonov A, Abu-Khalaf MM, Bianchini G. New strategies in breast cancer: Immunotherapy. Clin Cancer Res 2016;22:2105-10.
|
| 10. |
Redig AJ, Jänne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol 2015;33:975-7.
|
| 11. |
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48-61.
|
| 12. |
Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008;133:775-87.
|
| 13. |
Merlo A, Casalini P, Carcangiu ML, Malventano C, Triulzi T, Mènard S, et al. FOXP3 expression and overall survival in breast cancer. J Clin Oncol 2009;27:1746-52.
|
| 14. |
Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanović S, Robbins PF, et al. Tumor-infiltrating human CD4+regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol 2019;4. pii: eaao4310.
|
| 15. |
Ceeraz S, Nowak EC, Noelle RJ. B7 family checkpoint regulators in immune regulation and disease. Trends Immunol 2013;34:556-63.
|
| 16. |
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015;27:450-61.
|
| 17. |
Jung K, Choi I. Emerging co-signaling networks in T cell immune regulation. Immune Netw 2013;13:184-93.
|
| 18. |
Tripathi S, Guleria I. Role of PD1/PDL1 pathway, and TH17 and treg cells in maternal tolerance to the fetus. Biomed J 2015;38:25-31.
[PUBMED] [Full text] |
| 19. |
Pentcheva-Hoang T, Corse E, Allison JP. Negative regulators of T-cell activation: Potential targets for therapeutic intervention in cancer, autoimmune disease, and persistent infections. Immunol Rev 2009;229:67-87.
|
| 20. |
Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev 2008;224:166-82.
|
| 21. |
Vonderheide RH, LoRusso PM, Khalil M, Gartner EM, Khaira D, Soulieres D, et al. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin Cancer Res 2010;16:3485-94.
|
| 22. |
McArthur HL, Diab A, Page DB, Yuan J, Solomon SB, Sacchini V, et al. Apilot study of preoperative single-dose ipilimumab and/or cryoablation in women with early-stage breast cancer with comprehensive immune profiling. Clin Cancer Res 2016;22:5729-37.
|
| 23. |
Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007;27:111-22.
|
| 24. |
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027-34.
|
| 25. |
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001;2:261-8.
|
| 26. |
Zinselmeyer BH, Heydari S, Sacristán C, Nayak D, Cammer M, Herz J, et al. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med 2013;210:757-74.
|
| 27. |
Honda T, Egen JG, Lämmermann T, Kastenmüller W, Torabi-Parizi P, Germain RN. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity 2014;40:235-47.
|
| 28. |
Vibhakar R, Juan G, Traganos F, Darzynkiewicz Z, Finger LR. Activation-induced expression of human programmed death-1 gene in T-lymphocytes. Exp Cell Res 1997;232:25-8.
|
| 29. |
Zielinski C, Knapp S, Mascaux C, Hirsch F. Rationale for targeting the immune system through checkpoint molecule blockade in the treatment of non-small-cell lung cancer. Ann Oncol 2013;24:1170-9.
|
| 30. |
Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, et al. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2015;6:5449-64.
|
| 31. |
Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A 2007;104:3360-5.
|
| 32. |
Muenst S, Soysal SD, Gao F, Obermann EC, Oertli D, Gillanders WE. The presence of programmed death 1 (PD-1)-positive tumor-infiltrating lymphocytes is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat 2013;139:667-76.
|
| 33. |
Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res 2014;2:361-70.
|
| 34. |
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002;99:12293-7.
|
| 35. |
Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol 2018;200:422-31.
|
| 36. |
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320-30.
|
| 37. |
Chesney JA, Mitchell RA, Yaddanapudi K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J Leukoc Biol 2017;102:727-40.
|
| 38. |
Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med 2009;360:790-800.
|
| 39. |
Reis-Filho JS, Weigelt B, Fumagalli D, Sotiriou C. Molecular profiling: Moving away from tumor philately. Sci Transl Med 2010;2:47ps43.
|
| 40. |
Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 2008;14:518-27.
|
| 41. |
Ignatiadis M, Singhal SK, Desmedt C, Haibe-Kains B, Criscitiello C, Andre F, et al. Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: A pooled analysis. J Clin Oncol 2012;30:1996-2004.
|
| 42. |
Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res 2008;14:5158-65.
|
| 43. |
Schmidt M, Böhm D, von Törne C, Steiner E, Puhl A, Pilch H, et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res 2008;68:5405-13.
|
| 44. |
Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol 2007;8:R157.
|
| 45. |
Bianchini G, Qi Y, Alvarez RH, Iwamoto T, Coutant C, Ibrahim NK, et al. Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J Clin Oncol 2010;28:4316-23.
|
| 46. |
Callari M, Cappelletti V, D’Aiuto F, Musella V, Lembo A, Petel F, et al. Subtype-specific metagene-based prediction of outcome after neoadjuvant and adjuvant treatment in breast cancer. Clin Cancer Res 2016;22:337-45.
|
| 47. |
DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: Crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res 2007;9:212.
|
| 48. |
Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol 2015;33:983-91.
|
| 49. |
Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, et al. Tumor-infiltrating CD8+lymphocytes predict clinical outcome in breast cancer. J Clin Oncol 2011;29:1949-55.
|
| 50. |
Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proc Natl Acad Sci U S A 2012;109:2796-801.
|
| 51. |
Dedeurwaerder S, Desmedt C, Calonne E, Singhal SK, Haibe-Kains B, Defrance M, et al. DNA methylation profiling reveals a predominant immune component in breast cancers. EMBO Mol Med 2011;3:726-41.
|
| 52. |
Andre F, Job B, Dessen P, Tordai A, Michiels S, Liedtke C, et al. Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res 2009;15:441-51.
|
| 53. |
Desmedt C, Di Leo A, de Azambuja E, Larsimont D, Haibe-Kains B, Selleslags J, et al. Multifactorial approach to predicting resistance to anthracyclines. J Clin Oncol 2011;29:1578-86.
|
| 54. |
Issa-Nummer Y, Darb-Esfahani S, Loibl S, Kunz G, Nekljudova V, Schrader I, et al. Prospective validation of immunological infiltrate for prediction of response to neoadjuvant chemotherapy in HER2-negative breast cancer – A substudy of the neoadjuvant GeparQuinto trial. PLoS One 2013;8:e79775.
|
| 55. |
Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol 2008;8:59-73.
|
| 56. |
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: Immunostimulation by anticancer drugs. Nat Rev Drug Discov 2012;11:215-33.
|
| 57. |
Dieci MV, Mathieu MC, Guarneri V, Conte P, Delaloge S, Andre F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in two phase III randomized adjuvant breast cancer trials. Ann Oncol 2015;26:1698-704.
|
| 58. |
Sabatier R, Finetti P, Cervera N, Lambaudie E, Esterni B, Mamessier E, et al. Agene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res Treat 2011;126:407-20.
|
| 59. |
Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann Oncol 2015;26:259-71.
|
| 60. |
Mittendorf EA, Peoples GE, Singletary SE. Breast cancer vaccines: Promise for the future or pipe dream? Cancer 2007;110:1677-86.
|
| 61. |
DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009;16:91-102.
|
| 62. |
Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med 2012;18:1224-31.
|
| 63. |
Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol 2008;26:1789-96.
|
| 64. |
Tamura K, Shimizu C, Hojo T, Akashi-Tanaka S, Kinoshita T, Yonemori K, et al. FcγR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol 2011;22:1302-7.
|
| 65. |
Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010;18:485-98.
|
| 66. |
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 2017;16:2598-608.
|
| 67. |
Bonta II, Isac JF, Meiri E, Bonta D, Rich P. Correlation between tumor mutation burden and response to immunotherapy. ASCO 2017 annual meeting. J Clin Oncol (Chicago) 2017;35, abstract 15.
|
| 68. |
Barroso-Sousa R, Jain E, Kim D, Partridge AH, Cohen O, Wagle N. Determinants of high tumor mutational burden (TMB) and mutational signatures in breast cancer. Asco Annual Meeting (Chicago), J Clin Oncol 2018;36:15-6.
|
| 69. |
Xu J, Guo X, Jing M, Sun T. Prediction of tumor mutation burden in breast cancer based on the expression of ER, PR, HER-2, and Ki-67. Onco Targets Ther 2018;11:2269-75.
|
| 70. |
Gettinger S, Horn L, Jackman D, Spigel D, Antonia S, Hellmann M, et al. Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: Results from the CA209-003 study. J Clin Oncol 2018;36:1675-84.
|
| 71. |
Vokes EE, Ready N, Felip E, Horn L, Burgio MA, Antonia SJ, et al. Nivolumab versus docetaxel in previously treated advanced non-small-cell lung cancer (CheckMate 017 and checkMate 057): 3-year update and outcomes in patients with liver metastases. Ann Oncol 2018;29:959-65.
|
| 72. |
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803-13.
|
| 73. |
Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345-56.
|
| 74. |
Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, andin vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846-56.
|
| 75. |
Champiat S, Ferté C, Lebel-Binay S, Eggermont A, Soria JC. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. Oncoimmunology 2014;3:e27817.
|
| 76. |
Rosenberg SA. Decade in review-cancer immunotherapy: Entering the mainstream of cancer treatment. Nat Rev Clin Oncol 2014;11:630-2.
|
| 77. |
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509-20.
|
| 78. |
Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol 2013;31:860-7.
|
| 79. |
Ali HR, Glont SE, Blows FM, Provenzano E, Dawson SJ, Liu B, et al. PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann Oncol 2015;26:1488-93.
|
| 80. |
Wimberly H, Brown JR, Schalper K, Haack H, Silver MR, Nixon C, et al. PD-L1 expression correlates with tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy in breast cancer. Cancer Immunol Res 2015;3:326-32.
|
| 81. |
Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T (regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013;5:200ra116.
|
| 82. |
Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase ib KEYNOTE-012 study. J Clin Oncol 2016;34:2460-7.
|
| 83. |
Dirix LY, Takacs I, Jerusalem G, Arkenau HT, Hamilton EP, et al. Sint Augustinus. Avelumab (MSB0010718C), an anti-PD-L1 Antibody, in Patients with Locally Advanced or Metastatic Breast Cancer: A Phase Ib JAVELIN Solid Tumor Trial; Breast Cancer Res Treat 2018; 167(3):671-86.
|
| 84. |
Emens LA, Braiteh FS, Cassier P, DeLord JP, Eder JP, Shen XD, et al. Inhibition of PD-L1 by MPDL3280A leads to clinical activity in patients with metastatic triple-negative breast cancer. Cancer Res 2015;75:16:1010-24.
|
| 85. |
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889-94.
|
| 86. |
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23.
|
| 87. |
Persson J, Beyer I, Yumul R, Li Z, Kiem HP, Roffler S, et al. Immuno-therapy with anti-CTLA4 antibodies in tolerized and non-tolerized mouse tumor models. PLoS One 2011;6:e22303.
|
| 88. |
Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, Yagita H, et al. Anti-erbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci U S A 2011;108:7142-7.
|
| 89. |
Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010;18:160-70.
|
| 90. |
Kanjanapan YD, Wang L, Khalaf D, Yip S. ASCO annual meeting. J Clin Oncol (Chicago) 2018; (suppl; abstr 1029).
|
| 91. |
Hamm CA, Moran D, Rao K, Trusk PB, Pry K, Sausen M, et al. Genomic and immunological tumor profiling identifies targetable pathways and extensive CD8+/PDL1+immune infiltration in inflammatory breast cancer tumors. Mol Cancer Ther 2016;15:1746-56.
|
| 92. |
Bertucci F, Finetti P, Colpaert C, Mamessier E, Parizel M, Dirix L, et al. PDL1 expression in inflammatory breast cancer is frequent and predicts for the pathological response to chemotherapy. Oncotarget 2015;6:13506-19.
|
| 93. |
Recchia F. Maintenance immunotherapy in patients with metastatic breast cancer (MBC) who have a clinical benefit with chemotherapy. Long-term follow-up of a phase II study. ASCO annual meeting. J Clin Oncol (Chicago) 2018;36:abstract 15.
|
| 94. |
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med 2017;377:1919-29.
|
| 95. |
Criscitiello C. Tumor-associated antigens in breast cancer. Breast Care (Basel) 2012;7:262-6.
|
| 96. |
Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov 2013;3:388-98.
|
| 97. |
Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol 2003;200:290-7.
|
| 98. |
Globerson-Levin A, Waks T, Eshhar Z. Elimination of progressive mammary cancer by repeated administrations of chimeric antigen receptor-modified T cells. Mol Ther 2014;22:1029-38.
|
| 99. |
Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: Driving T cells to solid tumors. Cancer Discov 2016;6:133-46.
|
| 100. |
Li YR, Xian RR, Ziober A, Conejo-Garcia J, Perales-Puchalt A, June CH, et al. Mesothelin expression is associated with poor outcomes in breast cancer. Breast Cancer Res Treat 2014;147:675-84.
|
| 101. |
Tchou J, Wang LC, Selven B, Zhang H, Conejo-Garcia J, Borghaei H, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat 2012;133:799-804.
|
| 102. |
Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest 2016;126:3814-26.
|
| 103. |
Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012;119:4133-41.
|
| 104. |
Emens LA. Chemotherapy and tumor immunity: An unexpected collaboration. Front Biosci 2008;13:249-57.
|
| 105. |
Emens LA, Jaffee EM. Leveraging the activity of tumor vaccines with cytotoxic chemotherapy. Cancer Res 2005;65:8059-64.
|
| 106. |
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med 2018;378:2288-301.
|
| 107. |
Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med 2018;378:2078-92.
|
| 108. |
Arbour KC, Mezquita L, Long N, Rizvi H, Auclin E, Ni A. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non-small-cell lung cancer. J Clin Oncol 2018;36:2872-8.
|
| 109. |
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 2018;379:2108-21.
|
| 110. |
Pusztai L, Hofstatter EW, Chung GG, Horowitz NR, Lannin DR, Killelea BK. Durvalumab (MEDI4736) concurrent with nab-paclitaxel and dose dense doxorubicin cyclophosphamide (ddAC) as neoadjuvant therapy for triple negative breast cancer (TNBC). J Clin Oncol 2018;36:18.
|
| 111. |
Schmid P, Cortes J, Bergh JC, Pusztai L, Denkert C, Verma S. KEYNOTE-522: Phase III study of pembrolizumab (pembro) plus chemotherapy (chemo) vs. placebo plus chemo as neoadjuvant therapy followed by pembro vs. placebo as adjuvant therapy for triple-negative breast cancer (TNBC). Asco Annual Meeting (Chicago), J of Clinical Oncology 2018;36 suppl 15: TPS602.
|
| 112. |
Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol 2016;9:47.
|
| 113. |
Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res 2016;22:1499-509.
|