Peng Jiang, Wenjing Du, Xiaolu Yang
Department of Cancer Biology, Abramson Family Cancer Research Institute, Perelman School of Medicine, University of Pennsylvania, PA 19104, USA
| Date of Submission | 16-Aug-2013 |
| Date of Acceptance | 28-Sep-2013 |
| Date of Web Publication | 06-Dec-2013 |
Correspondence Address:
Xiaolu Yang
Department of Cancer Biology, Abramson Family Cancer Research Institute, Perelman School of Medicine, University of Pennsylvania, PA 19104
USA
Source of Support: None, Conflict of Interest: None
DOI: 10.4103/1477-3163.122760
Abstract
The tumor suppressor p53 plays a pre-eminent role in protecting against cancer, through its ability to sense various stresses and in turn invoke anti-proliferative and repair responses. Emerging evidence suggest that p53 is both a central sentinel for metabolic stresses and a master regulator of metabolic fluxes. This newly identified function of p53, along with the ability of p53 to induce senescence, appears to be crucial for the prevention of oncogenic transformation. A better understanding of the reciprocal regulation of p53 and metabolism, as well as p53-mediated connection between metabolism and senescence, may lead to the identification of valuable targets for tumor therapy.
Keywords: Metabolism, p53, senescence, tumor suppression
How to cite this article:
Jiang P, Du W, Yang X. p53 and regulation of tumor metabolism. J Carcinog 2013;12:21
How to cite this URL:
Jiang P, Du W, Yang X. p53 and regulation of tumor metabolism. J Carcinog [serial online] 2013 [cited 2021 Oct 13];12:21. Available from: https://carcinogenesis.com/text.asp?2013/12/1/21/122760
Introduction
The p53 protein is the central component of an elaborate tumor suppression network that monitors the well-being of the cell and determines cell fate. [1],[2] p53 responds to a myriad of severe stresses including deoxyribonucleic acid (DNA) damage, oncogene activation, telomere attrition and hypoxia, as well as low-level stresses that occur during normal physiological processes. In turn, p53 alters a variety of processes that prevent proliferation, repair damages, or optimize cell physiology. The importance of p53 in tumor suppression is underscored by the prevalence of p53 mutations, the single most common event in human tumors, which are present in 50-70% of tumors (http://p53.free.fr/). Even in tumors with a wild-type p53, the p53 pathway is frequently inactivated through alterations in its regulators or effectors. Inactivation of the p53 network is not only essential to tumor initiation, but also for their maintenance and progression. Nevertheless, how exactly p53 prevents tumor formation is not yet well-understood.
However, p53 is known to induce a range of anti-proliferative processes, specifically cell cycle arrest, apoptosis and senescence in response to stresses. Also p53 plays a critical role in monitoring and modulating the metabolic state of the cell. [3] Emerging evidence suggest that the ability of p53 to induce senescence may be crucial for suppression of oncogene-induced tumor formation. [4],[5],[6],[7],[8] Moreover, a recent study on mice that express a mutant form of p53 defective in acetylation, which cannot effectively induce cell cycle arrest, apoptosis, or senescence, shows that spontaneous tumor formation is still largely prevented. [9] Of note, this mutant p53 appears to retain the ability to modulate metabolism, suggesting that p53-mediated metabolic regulation may also be important for tumor suppression. How p53 modulates cellular metabolism and how it may link metabolism alteration with senescence induction are exciting areas of investigation.
P53 and Glucose Metabolism
Metabolism of tumor cells is markedly different from that of normal cells, which are mostly quiescent. A main nutrient for mammalian cells is glucose. As noted in the 1920s by Otto Warburg, tumor cells consume a much larger amount of glucose compared with their normal counterparts, and they rapidly generate lactate, even in the presence of an adequate oxygen, owing to accelerated glycolysis (aerobic glycolysis or the Warburg effect). [10],[11] The Warburg effect is also observed in rapidly dividing normal cells, indicating that it likely represents a metabolic adaptation to support cell proliferation rather than a defect of tumor cells.
The precise benefits of the Warburg effect to proliferating cells are still a subject of an intensive investigation, but likely include robust and highly adjustable subsidiary metabolic fluxes for the generation of amino acids, lipids and nucleotides that are required for cell growth. [12],[13],[14],[15] The Warburg effect may also help prevent the accumulation of reactive oxygen species (ROS) and minimize their damages to cellular constituents. This occurs through the reduced use of the electron transport chain, a main source of ROS and enhanced production of reduced nicotinamide adenine dinucleotide phosphate (NADPH), a reducing equivalent crucial for ROS detoxification. Moreover, maximal cell proliferation likely demands not only rapid production of adenosine tri-phosphate (ATP), NADPH and other metabolites required for biosynthesis and ROS detoxification, but also a proper balance among them that is in line with the constituents of the cell and the energy required for making them. Glycolysis, albeit a less efficient pathway in terms of the number of ATP generated per glucose molecule, enables fast ATP production and, at the same time, may help balance the supply of these metabolites.
While in normal cells the metabolic re-programming is likely orchestrated by mitogenic signals and highly regulated, in tumor cells it is brought about through mutations in oncogenes, tumor suppressor genes and in some cases, metabolic genes themselves. A number of oncogenes (e.g., Ras, phosphatidylinositol 3Ͳ-kinase/AKT, and hypoxia-inducible factor) were previously found to stimulate glucose metabolism. Recent studies indicated that p53 plays a major role in suppressing glucose consumption and antagonizing the Warburg effect. p53 modulates glucose consumption and glycolysis at multiple levels. Glucose transporters facilitate the uptake of glucose [Figure 1]. Among these transporters, GLUT1 and GLUT3 are expressed in most mammalian cells and have high affinity for glucose. p53 reduces the expression of GLUT3 through the inhibition of the IKK/nuclear factor-kB pathway, which stimulates GLUT3 expression. [16] p53 also directly suppresses the expression of GLUT1 and GLUT4, an insulin-regulated glucose transporter. [17] The loss of these functions of p53 leads to enhanced glucose consumption.
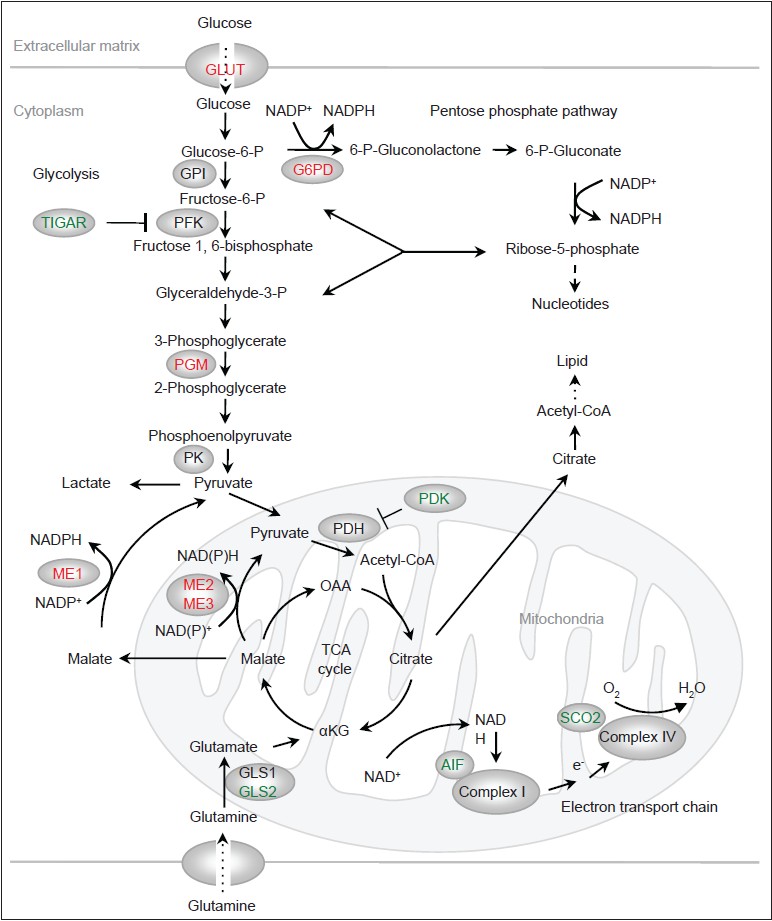 |
Figure 1: Regulation of metabolism by p53. Proteins that are activated by p53 are indicated in green and proteins that are inhibited by p53 are indicated in red. OAA: Oxaloacetate. α KG: α -ketoglutarate Click here to view |
The committing step of glycolysis is catalyzed by phosphofructokinase 1 (PFK1), which converts fructose 6-phosphate to fructose 1,6-bisphosphate [Figure 1]. PFK1 is the most important control site for the glycolytic flux, inhibited by ATP and citrate and activated by AMP. [18] Another important activator of PFK1 is fructose 2,6-bisphosphate, a metabolite whose levels are controlled by the bi-functional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK2/FBPase). p53 induces the expression of TP53-induced glycolysis and apoptosis regulator (TIGAR), which exhibits sequence similarity to the bisphosphatase2 domain of PFK2/FBPase and is able to reduce levels of fructose 2,6-bisphosphate. [19] Through the up-regulation of TIGAR, p53 lowers the glycolytic rate. Of note, TIGAR can also be expressed independently of p53, which occurs in a number of tumor cells, and it plays a role in tumorigenesis. [20]
Phosphoglycerate mutase (PGM) acts at the middle stage of glycolysis, converting 3-phosphoglycerate to 2-phosphoglycerate [Figure 1]. p53 reduces the levels of the PGM. [21] Interestingly, p53 does not affect the expression of the PGM genes; rather, it promotes the degradation of PGM proteins through an undefined mechanism. Nevertheless, both the outcome and mechanism of p53-mediated regulation of PGM appear to be cell type-specific. While p53 reduces the protein levels of PGM in embryonic fibroblast cells, it stimulates the expression of the PGM gene in muscle cells. [22]
In addition to generating pyruvate via glycolysis, glucose, in the form of glucose-6-phosphate, is shunted to the pentose phosphate pathway (PPP) [Figure 1]. The PPP consists of two branches: (1) An irreversible, oxidative branch where ribose-5-phosphate (R5P) is generated with the concomitant production of two NADPH molecules per glucose; and (2) a reversible, non-oxidative branch where R5P is inter-converted with the glycolytic intermediates fructose-6-phosphate and glyceraldehyde 3-phosphate. R5P is an obligatory precursor for de novo synthesis of nucleotides as well as other metabolites including NAD (P) + and ATP. NADPH, in addition to being the “reducing power” for several anti-oxidant systems, also supplies electrons for reductive biosynthesis including the production of lipids, deoxynucleotides and cholesterol. [23],[24],[25]
The pace of the oxidative PPP is set by glucose-6-phosphate dehydrogenase (G6PD), the first enzyme of this pathway. Interestingly, p53 inhibits G6PD and this function of p53 is observed in various cell lines and mouse tissues. [26] p53 deficient cells shows an increased PPP flux, NADPH production and glucose consumption, which can be largely reversed via the inhibition of G6PD. This suggests that p53 inactivation contributes to avid glucose consumption, an important aspect of the Warburg effect, through a hyperactive G6PD. Of note, p53 does not affect the expression of the G6PD gene or the stability of the G6PD protein. Instead, it binds to the G6PD protein and converts it from an active dimer to an inactive monomer. Moreover, p53 can inactivate G6PD at a sub-stoichiometric ratio through transient interaction, suggesting a “catalytic” activity of p53 in facilitating protein conformational changes. [26] It would be interesting to determine the biochemical basis of this previously-unrecognized function of p53 and whether similar mechanisms may underlie the ability of p53 in modulating proteins involved in apoptosis and other processes.
Induction of TIGAR and perhaps, also inactivation of PGM mediated by p53 lead to accumulation of glucose-6-phosphate, which indirectly raises the PPP flux. However, because the net result of p53 inactivation is an enhanced, instead of a decreased PPP flux, [26] p53-mediated inhibition of G6PD likely overrides the effect of TIGAR. Nevertheless, the inhibition of G6PD through protein-protein interactions, which likely occurs rapidly, in combination with the transcriptional up-regulation of TIGAR, which is expected to occur at relatively low kinetics, may enable fine-tuned, temporal regulation of the PPP by p53.
The PPP is enhanced in tumor cells through other direct and indirect mechanisms. For example, pyruvate kinase (PK)-M2, an isoform of PK that catalyzes the conversion of phosphoenol pyruvate to pyruvate, is widely expressed in tumor cells. Because PK-M2 has a relatively weak activity compared with the other PK isoforms, its expression leads to build-up of glycolytic intermediates to be shunted to the PPP. [27] The activity of PK-M2 is reduced under conditions of high ROS, leading to a further enhancement of the PPP. [28] The ataxia-telangiectasia mutated kinase (ATM), which orchestrates DNA damage responses, enhances the activity of G6PD through the phosphorylation of HSP27, an activator or G6PD. [29] More recently, TAp73, the transcriptional competent isoform of the p53 family protein p73, was shown to directly stimulate the expression of the G6PD genes. [30] TAp73, unlike p53, is rarely mutated in human tumor cells. The regulation of G6PD by TAp73 indicates a role of TAp73 in cell proliferation and further highlights the link between the PPP and oncogenic growth.
Glutamine Metabolism
Glutamine, along with glucose, supplies cells with most of the free energy, biosynthetic precursors and reducing equivalents that are necessary for growth and proliferation. [31],[32],[33] Many tumor cells avidly consume glutamine and convert it to pyruvate or lactate (glutaminolysis). [34] Like glycolysis, a robust glutaminolysis may enable glutamine flux into subsidiary pathways for biosynthesis. Moreover, glutaminolysis itself generates ATP and NADPH. In glycolytic cells, glucose-derived pyruvate is diverted from the mitochondria and tricarboxylic acid cycle (TCA cycle) intermediates are extruded for biosynthesis. In these cells, glutamine plays a major role in replenishing the TCA intermediates (anaplerosis). In addition, the re-entry of glutamine-derived pyruvate to the TCA cycle allows for glutamine to be used as a substrate for oxidative phosphorylation and ATP production. Moreover, in tumor cells growing under hypoxic conditions or in tumor cells harboring mutations in the TCA cycle or the electron transport chain, mitochondria may cease to function properly. Interestingly, in these cells glutamine-derived α-ketoglutarate undergoes reductive carboxylation to generate isocitrate, which is then isomerized to citrate and onwards for fatty acid synthesis, maintaining tumor cell growth. [35],[36],[37]
The oncoprotein c-Myc plays a significant role in stimulating glutamine metabolism through the up-regulation of various genes involved in this process. [38],[39],[40] Recent studies also revealed a complex role of p53 in regulating glutamine metabolism. Glutamine is converted by the glutaminase to glutamate [Figure 1]. Glutamate can turn into α-ketoglutarate and enter the TCA cycle, or be incorporated into glutathione (GSH), an abundant cellular anti-oxidant that plays a major role in repairing oxidatively damaged proteins. p53 strongly stimulates the expression of the glutaminase isoform GLS2, leading to an increase in cellular GSH levels and reduction in ROS. [41],[42] Up-regulation of GLS2 is found in tumor cells, [43] and enforced expression of GLS2 inhibits tumor cell growth. [41],[42] Paradoxically, c-Myc stimulates the expression of the other glutaminase isoform, GLS1. [40] Inhibition of GLS1 by small compounds suppresses, rather than enhances, oncogenic growth. [44] The reason for the opposing effects of GLS1 and GLS2 on tumor cell growth is not understood.
A critical step in glutamine metabolism is the conversion of malate to pyruvate, which enables both glutaminolysis and the use of glutamine as a TCA substrate. This step is catalyzed by malic enzymes, with the concomitant generation of either NADPH or NADH [Figure 1]. [45],[46] There are three malic enzyme isoforms in mammalian cells that differ in sub-cellular localizations and co-factor preferences: (1) a cytoplasmic, NADP + -dependent isoform (ME1); (2) a mitochondrial, NADP + or NAD + -dependent isoform (ME2); and (3) a mitochondrial, NADP + -dependent isoform (ME3), with ME1 and ME2 being the major isoforms in a number of cell lines. Both ME1 and ME2 strongly influence glutamine consumption while having a moderate effect on glucose consumption. [47] These enzymes also regulate cellular NADPH and ROS levels and the production of lipids, [47] with either ME1 or ME2 playing a more dominant role in these processes depending on cell type. Interestingly, p53 suppresses the expression of all ME isoforms through binding to consensus response elements within these genes. Through the inhibition of malic enzymes, p53 regulates glutamine metabolism, NAPDH production, redox state and biosynthesis. [47]
The TCA Cycle, Oxidative Phosphorylation and Lipid Oxidation
The TCA cycle is a central metabolic hub for bioenergetics (through its connection with electron transport chain and oxidative phosphorylation) and for biosynthesis (by providing precursors for the production of lipids, nucleotides and amino acids). Glucose- and glutamine-derived pyruvate becomes the TCA cycle substrate acetyl CoA through a reaction catalyzed by the pyruvate dehydrogenase (PDH) complex [Figure 1]. Acetyl-CoA is then condensed with oxaloacetate to become the TCA intermediate citrate. PDH is inactivated through phosphorylation mediated by pyruvate dehydrogenase kinase (PDK) and is activated through de-phosphorylation mediated by pyruvate dehydrogenase phosphatase. A recent study showed that p53 suppresses the expression of PDK2, which may enhance pyruvate flux into the TCA cycle. [48]
Oxidative phosphorylation is powered by the mitochondrial membrane potential generated through the action of a set of proton translocating complexes of the electron transport chain. p53 activates the expression of the synthesis of cytochrome c oxidase 2 (SCO2), which facilitates the assembly of complex IV (cytochrome c oxidase). [49] It also transcriptionally enhances apoptosis-inducing factor (AIF), [50] a mitochondrial pro-apoptotic protein that is also required for the function of complex I (NADH dehydrogenase). [51] Thus, p53 helps shift the production of ATP from glycolysis to oxidative phosphorylation.
p 53 -0 Mediated Connection Between Metabolism and Senescence
Given the role of p53 as a remarkably versatile stress sensor and its powerful effect it has on cells, it is perhaps not surprising that p53 can also be activated in response to fluctuations in nutrient availability and metabolic fluxes, with consequences ranging from adjusting metabolism, enhancing cell survival, to inducing apoptosis/senescence. p53 is activated upon glucose deprivation in a manner that is dependent on the AMP-activated kinase (AMPK) complex, which coordinates cellular response to energy availability. [52] This AMPK-p53 pathway likely constitutes a metabolic checkpoint. Of note, activation of p53 under glucose deprivation enhances cell survival through the induction of temporal cell cycle arrest. p53 is also activated under conditions of serine deprivation. [53] Activation of p53 and the p53-target gene p21 leads to transient cell cycle arrest, allowing for reduced intracellular pool of serine to be used for GSH production and maintaining a favorite cellular redox state. The pro-survival effect of p53 may be also related to its ability to promote mitochondrial fatty acid oxidation [54] and to induce autophagy, [55],[56] thereby providing alternative energy sources for starved cells. These findings have implications in treating p53-deficient tumors through limiting the relevant nutrient or the controlled activation of the relevant pathway (e.g., the AMPK pathway). [53],[54] Nevertheless, the effect of p53 on cell fate is likely determined by the duration and severity of metabolic stresses, as prolonged glucose deprivation results in senescence in p53 wild-type cells. [52]
p53 is strongly activated by even relatively mild down-regulation of ME1 or ME2, while enforced expression of these enzymes delays p53 activation, [47] suggesting that p53 also acts as a sensor for the ME flux. Interestingly, ME1 and ME2 affect p53 through distinct mechanisms. Down-regulation of ME1 activates p53 through a reduction in the expression of the Mdm2 gene, which encodes the principal E3 ligase for p53, while down-regulation of ME2 activates p53 due to elevation in ROS levels, which turns on the p53 activator AMPK. Thus, p53 and malic enzymes form a positive feedback loop that bolsters p53 activation. Of note, the consequence of the p53 activation is the induction of senescence, not apoptosis and this occurs even in tumor cells. Thus, this feedback loop selectively influences the outcome of p53 activation.
p53-regulated glycolytic enzyme PGM can immortalize primary mouse fibroblast cells upon over-expression. So can another glycolytic enzyme, glucose-6-phosphate isomerase (GPI), which catalyzes the conversion of glucose-6-phosphate into fructose 6-phosphate. Conversely, knockdown of PGM and GPI results in senescence. [21] Furthermore, an increase in glycolytic flux is observed in immortalized and stem cells, [21] suggesting a link between a robust glycolysis and the suppression of replicative senescence. In addition, enforced expression of G6PD is shown to transform immortalized cells, [57] while knockdown of G6PD can induce senescence. [30] The PDK complex is critical for oncogene-induced senescence and enhanced use of pyruvate in the TCA cycle is associated with higher oxidative stress. [58] Thus, p53 may also promote senescence via the suppression of glycolysis and the PPP, or the increase in the use of pyruvate in the TCA cycle. Again, this effect of p53 may be related to the extent of p53 activation, which is in turn influenced by metabolic conditions such as glucose and serine availability as well as malic enzyme fluxes. The diverse consequences of p53 activation in response to metabolic stresses underscore the role of p53 as both an optimizer of cell physiology and an arbiter of cell fate.
In summary, although the pre-eminent role of p53 in protecting again cancer is firmly established, the underlying mechanism remains unclear and is an exciting, yet challenging area of research. Given that inactivation of p53 is required for both the initiation and progression of a wide variety of tumor cells, re-activation of p53 in p53-wild type tumor cells, or blockage of critical tumor-promoting pathways that become hyperactive in p53-deficient tumors, is a highly attractive method to therapeutically re-establish the growth-inhibitory functions of p53 in cancer cells. Future studies will improve our understanding of the importance of p53-induced metabolic alterations and senescence induction in tumor suppression, the connection between them and the reciprocal regulation of p53 by metabolic enzymes. Ultimately, this knowledge will likely lead to the identification of p53-regulating, as well as p53-regulated, enzymes which can serve as potential targets for restoring p53 function in tumor cells.
Acknowledgment
The author acknowledges supports from US National Institutes of Health (CA088868 and GM060911) and the Department of Defense (W81XWH-10-1-0468 and W81XWH-13-1-0100).
References
| 1. | Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000;408:307-10.  |
| 2. | Vousden KH, Prives C. Blinded by the Light: The growing complexity of p53. Cell 2009;137:413-31. |
| 3. | Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer 2009;9:691-700. |
| 4. | Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005;436:660-5. |
| 5. | Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005;436:725-30. |
| 6. | Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007;445:656-60. |
| 7. | Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007;445:661-5. |
| 8. | Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011;145:571-83. |
| 9. | Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012;149:1269-83. |
| 10. | Warburg O, Posener K, Negelein E. Ueber den stoffwechsel der tumoren. Biochem Z 1924;152:319-44. |
| 11. | Warburg O. On the origin of cancer cells. Science 1956;123:309-14. |
| 12. | Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009;324:1029-33. |
| 13. | Lunt SY, Vander Heiden MG. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011;27:441-64. |
| 14. | Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011;11:85-95. |
| 15. | Dang CV. Links between metabolism and cancer. Genes Dev 2012;26:877-90. |
| 16. | Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol 2008;10:611-8. |
| 17. | Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res 2004;64:2627-33. |
| 18. | Sola-Penna M, Da Silva D, Coelho WS, Marinho-Carvalho MM, Zancan P. Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life 2010;62:791-6. |
| 19. | Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006;126:107-20. |
| 20. | Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, et al. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev Cell 2013;25:463-77. |
| 21. | Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res 2005;65:177-85. |
| 22. | Ruiz-Lozano P, Hixon ML, Wagner MW, Flores AI, Ikawa S, Baldwin AS Jr, et al. p53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ 1999;10:295-306. |
| 23. | Berg JM, Tymoczko JL, Stryer L. Biochemistry 2007; 6 th ed: 577-89. |
| 24. | Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic Biol Med 2012;53:421-36. |
| 25. | Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012;64:362-9. |
| 26. | Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 2011;13:310-6. |
| 27. | Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008;452:230-3. |
| 28. | Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011;334:1278-83. |
| 29. | Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J 2011;30:546-55. |
| 30. | Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, et al. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol 2013;15:991-1000. |
| 31. | Newsholme EA, Crabtree B, Ardawi MS. Glutamine metabolism in lymphocytes: Its biochemical, physiological and clinical importance. Q J Exp Physiol 1985;70:473-89. |
| 32. | Baggetto LG. Deviant energetic metabolism of glycolytic cancer cells. Biochimie 1992;74:959-74. |
| 33. | DeBerardinis RJ, Cheng T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010;29:313-24. |
| 34. | Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 1979;254:2669-76. |
| 35. | Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A 2011;108:19611-6. |
| 36. | Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011;481:380-4. |
| 37. | Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011;481:385-8. |
| 38. | Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 2007;178:93-105. |
| 39. | Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008;105:18782-7. |
| 40. | Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009;458:762-5. |
| 41. | Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A 2010;107:7455-60. |
| 42. | Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A 2010;107:7461-6. |
| 43. | Gómez-Fabre PM, Aledo JC, Del Castillo-Olivares A, Alonso FJ, Núñez De Castro I, Campos JA, et al. Molecular cloning, sequencing and expression studies of the human breast cancer cell glutaminase. Biochem J 2000;345 Pt 2:365-75. |
| 44. | Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010;18:207-19. |
| 45. | Hsu RY. Pigeon liver malic enzyme. Mol Cell Biochem 1982;43:3-26. |
| 46. | Chang GG, Tong L. Structure and function of malic enzymes, a new class of oxidative decarboxylases. Biochemistry 2003;42:12721-33. |
| 47. | Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013;493:689-93. |
| 48. | Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res 2012;72:560-7. |
| 49. | Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science 2006;312:1650-3. |
| 50. | Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, et al. Regulation of AIF expression by p53. Cell Death Differ 2006;13:2140-9. |
| 51. | Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, et al. AIF deficiency compromises oxidative phosphorylation. EMBO J 2004;23:4679-89. |
| 52. | Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005;18:283-93. |
| 53. | Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013;493:542-6. |
| 54. | Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 2007;67:6745-52. |
| 55. | Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006;126:121-34. |
| 56. | Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008;10:676-87. |
| 57. | Kuo W, Lin J, Tang TK. Human glucose-6-phosphate dehydrogenase (G6PD) gene transforms NIH 3T3 cells and induces tumors in nude mice. Int J Cancer 2000;85:857-64. |
| 58. | Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013;498:109-12. |