Heidrun Weidemann
Department of Oncology, Hadassah-Hebrew University, Medical Center, Jerusalem, Israel
| Date of Submission | 18-Aug-2011 |
| Date of Acceptance | 01-Dec-2011 |
| Date of Web Publication | 17-Feb-2012 |
Correspondence Address:
Heidrun Weidemann
Department of Oncology, Hadassah-Hebrew University, Medical Center, Jerusalem
Israel
Source of Support: None, Conflict of Interest: None
Abstract
Since their first discovery as potential anti-cancer drugs decades ago, there is increasing evidence that digitalis-like compounds (DLC) have anti-tumor effects. Less is known about endogenous DLC (EDLC) metabolism and regulation. As stress hormones synthesized in and secreted from the adrenal gland, they likely take part in the hypothalamo-pituitary-adrenal (HPA) axis. In a previous study, we revealed reduced EDLC concentrations in plasma and organs from immune-compromised animals and proposed that a similar situation of a deregulated HPA axis with “adrenal EDLF exhaustion” may contribute to tumorigenesis in chronic stress situations. Here, we put forward the hypothesis that a lowered EDLC response threshold of tumor cells as compared with normal cells increases the risk of tumorigenesis, especially in those individuals with reduced EDLC plasma concentrations after chronic stress exposure. We will evaluate this hypothesis by (a) summarizing the effects of different DLC concentrations on tumor as compared with normal cells and (b) reviewing some essential differences in the Na/K-ATPase of tumor as compared with normal cells (isoform pattern, pump activity, mutations of other signalosome receptors). We will conclude that (1) tumor cells, indeed, seem to have their individual “physiologic” EDLC response range that already starts at pmolar levels and (2) that individuals with markedly reduced (pmolar) EDLC plasma levels are predisposed to cancer because these EDLC concentrations will predominantly stimulate the proliferation of tumor cells. Finally, we will summarize preliminary results from our department supporting this hypothesis.
Keywords: Adrenal exhaustion, endogenous digitalis-like compounds, isoforms, Na/K-ATPase, stress, threshold, tumorigenesis
How to cite this article:
Weidemann H. “The Lower Threshold” phenomenon in tumor cells toward endogenous digitalis-like compounds: Responsible for tumorigenesis?. J Carcinog 2012;11:2
How to cite this URL:
Weidemann H. “The Lower Threshold” phenomenon in tumor cells toward endogenous digitalis-like compounds: Responsible for tumorigenesis?. J Carcinog [serial online] 2012 [cited 2021 Oct 14];11:2. Available from: https://carcinogenesis.com/text.asp?2012/11/1/2/92999
Introduction
The endogenous digitalis-like compounds (EDLC) belong to a family of steroid hormones, which originally stem from plants and recently have been demonstrated to be synthesized and released mainly in the adrenal gland of different species. [1],[2],[3],[4],[5],[6],[7],[8],[9],[10] As “stress hormones” similar to cortisol, they are integrated in the feedback loops of the hypothalamic-pituitary-adrenal (HPA) axis and stimulated by ACTH and Angiotensin II. [11],[12],[13],[14],[15] The EDLC are the natural ligands of the Na/K-ATPase (NKA), the classical sodium pump. [16],[17],[18],[19] It is nowadays well established that the NKA represents also a signal transducer that is partly independent from its pump activity. [20],[21],[22],[23],[24],[25],[26],[27],[28],[29],[30] Since their first discovery as potential anti-cancer drugs decades ago, [31],[32],[33],[34] there is increasing evidence over the last years, in-vitro and in-vivo, that digitalis-like compounds (DLC) have anti-tumor properties. [35],[36],[37],[38],[39],[40],[41],[42],[43],[44],[45],[46] These include, for example induction of apoptosis via Ca 2+ -dependant caspase-3 activation, [47] promotion of cell cycle arrest and cell differentiation via upregulation of the cell cycle inhibitor p21 cip1 , [48] cell growth inhibition via downregulation of the NKA-α1 isoform, [49] NF-κB, [50] HIF-1α[51] as well as inhibition of topoisomerase I and II [52] and autophagic cell death via lysosomal membrane permeabilization [53] with interruption of the actin cytoskeleton. Remarkably, inhibition of the NKA by DLC also has been shown to sensitize cancer cells toward anoikosis. [54] Recently, different DLC partly derived from plants (Oleandrin from Nerium oleander L.) [55],[56],[57],[58] and partly semi-synthetic (UNBS1450 derived from Calotropis procera) [50],[59],[60] entered Phase I trials in solid tumors with, so far, no toxicity. Less is known about the endogenous DLC metabolism and their regulation. As mentioned above, they are part of the HPA axis and thus also depend on the integrity of the thymus. A bidirectional relationship between the thymus and the HPA axis is well established, with mutual dependency in maturation and function. [61],[62],[63],[64] As demonstrated in a previous study, nude mice, traditionally used for tumor transplantation, did not only reveal reduced basal EDLC concentrations in the adrenal gland but also did not respond to an acute stress stimulus, even showing decreased plasma EDLC concentrations after additional ACTH application. [15] We proposed that this “adrenal EDLC exhaustion” with gradually decreasing EDLC plasma concentrations in immune-compromised individuals, e.g. after chronic stress exposure, may contribute to tumorigenesis. [65] In the recent years, a lot of evidence has accumulated that exogenous and endogenous DLC are able to induce MAPK signaling pathways via the NKA/Src/epidermal growth factor receptor (EGFR) “signalosome” [66],[67],[68] and, hence, lead either to stimulated cell growth (hypertrophy), cell cycle arrest and cell differentiation, or apoptosis.
The kind of interaction between DLC and MAPK signaling pathways is dose- and time dependant, and, moreover, depends critically on the nature of the involved cell membrane receptors, especially the molecular structure (isozymes), activity and cellular amount of the NKA. It also depends on the mutation status of the tyrosine kinase receptors, e.g. the EGF-R.
We put forward the hypothesis that a lowered endogenous DLC response threshold of tumor as compared with normal cells [Figure 1] increases the risk of tumorigenesis, especially in individuals with reduced EDLC plasma concentrations after long stress exposure.
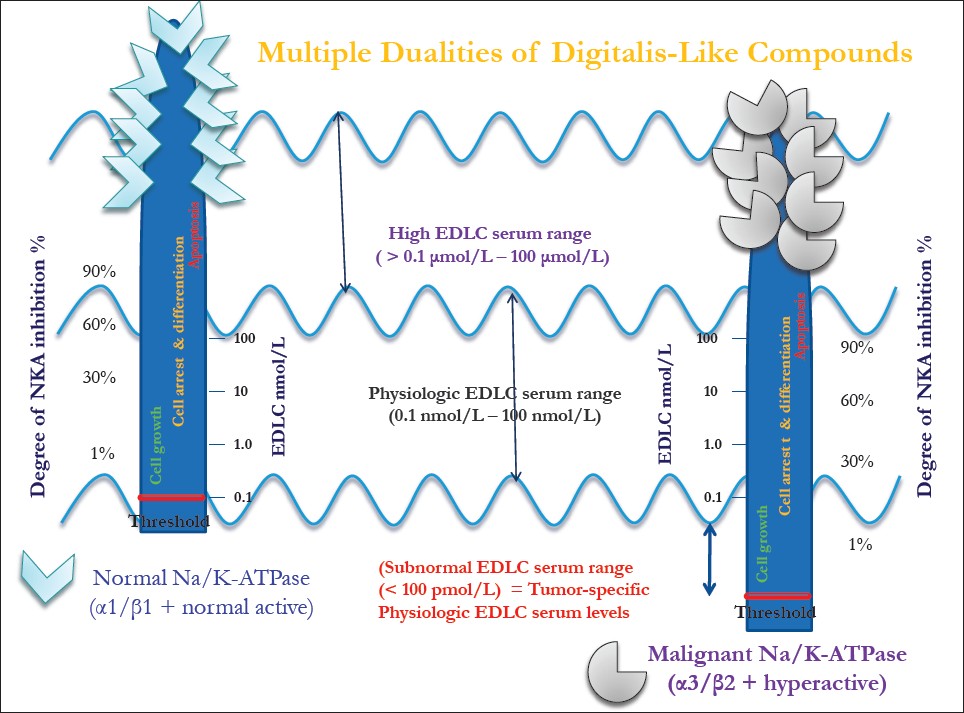 |
Figure 1: The lower-threshold theory in cancer. The dual effects of endogenous digitalis-like compounds (EDLC) are demonstrated, which are, in principle, similar for normal and tumor cells, with the only crucial difference being that the response threshold of the tumor cells toward EDLC shifted to a lower (pmolar) EDLC range Click here to view |
In the first part of evaluation, we will summarize data about either cell growth-stimulating or -inhibiting effects of DLC at different dosages on normal as well as tumor cells.
In the second part, we will recapitulate knowledge about the characteristics of the NKA in tumor as compared with normal cells. Finally, we will develop our hypothesis referring to the data mentioned before.
Summary of the Effects of Digitalis-Like Compounds
Sub-physiologic EDLC plasma concentrations
(1 pM-100 pM)
The in vitro studies analyzing the effect of DLC on diverse cell lines scarcely used these low DLC (pmolar) concentrations. Most of the studies start cell treatments at 1 nM-10 nM or 1 nM-100 nM. For instance, Qiu et al.[69] did not see any significant impact on cell growth of human umbilical vein endothelial cells (HUVEC) at ouabain concentrations <0.1 nm, i.e. neither cell proliferation nor apoptosis. To our knowledge, no data exist about the effects of pmolar DLC concentrations on malignant cells.
Physiologic EDLC plasma concentrations
(0.1 nM-10 nM; max., 100 nM)
Effect on normal cells
Qiu et al. [69] exposed HUVEC to different concentrations (0.1 nM-100 nM) of ouabain at 12-48-h intervals. Ouabain stimulated HUVEC cell proliferation at low concentrations (1.0 nM) and induced cell death at markedly higher (>100 nM) concentrations. Aydemir-Koksov et al.[70] demonstrated that ouabain at concentrations below those that inhibit the pump, i.e. 0.1 nM and 1.0 nM, induced trans-activation of the EGF-R, resulting in increased proliferation and bromodeoxyuridine (BrdUrd) uptake of canine vascular smooth muscle (VSMC) cells. Interestingly, higher ouabain concentrations (10 nM) had little or no stimulating effect on proliferation. Winnicka et al. [71] showed that in human fibroblasts, 30 nM ouabain, digoxin and proscillaridin A induced an anti-apoptotic action by increasing the level of phosphorylated extracellular signal-regulated kinases (p-ERK 1/2). Similarly, Chueh et al. [72] demonstrated that ouabain at low nM concentrations promoted cell proliferation in human prostate smooth muscle cells via a Ca(2+)-dependent mechanism and activation of the MEK-p42/44 MAPK pathway. Li et al.[73] investigated the effect of low-dose ouabain on the viability of rat renal proximal tubular cells. Ouabain (0.1 nM-10 nM) stimulated the proliferation of kidney cells; interestingly, these effects were abolished when slow calcium oscillations via the IP3R were prevented. Khundmiri et al. [74] observed that ouabain induced cell proliferation in opossium kidney tubular cells involving calcium-dependant phosphorylation of Akt. This effect started at ouabain 1 nM and was maximal at 10 nM-100 nM, whereas at 1 μM, ouabain decreased Akt-phosphorylation. Wei et al. [75] revealed that spiral ganglion neurons exposed to neurobasal medium + 10 nM ouabain had a much lower apoptosis index, increased Bcl-2 levels and, interestingly, longer dendrite growth as compared with neurons exposed to neurobasal medium only. De Rezende Correra et al. [76] demonstrated that ouabain significantly increased retinal ganglion cell survival, with a maximum at 3.0 nM, after 48 h in culture. The blockade of protein kinase C activity abolished the ouabain effect.
Effect on malignant cells
Lopez-Lazaro et al. [77] revealed that in three human cancer cell lines – TK-10 (renal), MCF-7 (breast) and UACC-62 (melanoma) – the IC50 values for digitoxin (3 nM-33 nM) were within the concentration range (20 nM-33 nM) seen in the plasma of patients with cardiac disease receiving this glycoside. Specifically, digitoxin at 1 in 30 nM induced levels of DNA-topoisomerase II cleavable complexes similar to etoposide. Kometiani et al. [78] explored the mechanism of the growth inhibitory effects of DLC on the estrogen receptor-negative human breast cancer cell line MDA-MB-435. Ouabain concentrations (10 nM and 100 nM) that caused less than 25% inhibition of the NKA pumping function activated Src kinase, stimulated the interaction of Src and Na/K-ATPase with EGF-R, caused a transient and then a sustained activation of ERK1/2 and increased the expression of the cell cycle inhibitor p21 Cip1 . Winnicka et al. [79] observed reduced cell viability in another human breast cancer cell line (MDA-MB-231) after applying ouabain, digoxin and proscillaridin A in nmol ranges. They confirmed that cardenolides induce apoptosis in MDA-MB-231 cells by increasing free calcium concentration and by activating caspase-3. Notably, they revealed marked differences in the potency, with proscillaridin A being the most active (IC50 48 ± 2 nM). Bielawski et al. [52] evaluated the role of cardenolides in MCF-7 breast cancer cells with special focus on topoisomerases. Both digoxin and ouabain inhibited topoisomerase II catalytic activity at 100 nM. Proscillaridin A was an even more potent poison of topoisomerase I and II activity at 30 nM and 100 nM, respectively. Mijatovic/Kiss et al. [53] developed a semi-synthetic cardenolide, UNBS1450, with a markedly higher affinity to the NKA of diverse human cell lines than ouabain or digoxin. UNBS1450 at 10 nM-100 nM induced in A549 NSCLC cells a cell death process associated with dramatic cytoplasmic vacuolization due to increased lysosomal permeabilization. Remarkably, neither signs of apoptotic nor necrotic cell death were seen. McConkey et al. [47] demonstrated in two variants (Pro4 and LN4) of the human prostate androgen-independent metastatic adenocarcinoma PC3 pro-apoptotic effects of cardiac glycosides (oleandrin, ouabain and digoxin). Concentration-response analyses revealed in both cell lines maximal responses of ouabain and digoxin at 100 nM. Huang et al. [80] also examined the cytotoxic effects of ouabain on the human prostate cancer cell line PC3. Low concentrations of ouabain (<10 nM) induced the increase of Par-4 expression and sensitized the cells toward cytotoxicity. Higher ouabain concentrations (<100 nM) induced a significant and time-dependent loss of mitochondrial membrane potential (Deltapsim), a sustained production of reactive oxygen species (ROS) and a severe apoptotic reaction. Smith et al. [81] examined the relative abilities of oleandrin, ouabain and anvirzel to inhibit FGF-2 export from two human prostate cancer cell lines, DU145 and PC3. Oleandrin (0.1 ng/mL) produced a 45.7% inhibition of FGF-2 release from PC3 cells and a 49.9% inhibition from DU145 cells. Non-cytotoxic concentrations (100 ng/mL) of anvirzel produced a 51.9% and 30.8% inhibition of FGF-2 release, respectively, in the two cell lines. Li et al. [82] demonstrated that in a human gastric cancer cell line (MGC803), bufalin at 20 nmol/L induced M-phase cell cycle arrest, whereas at 80 nmol/L, it induced apoptosis via an increased Bax/Bcl-2 ratio and activated caspase-3. Remarkably, these distinct effects were correlated to a transient activation of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway and a complete inhibition, respectively. Xu et al. [83] analyzed the combined effects of ouabain (5 nM-1000 nM) and α1-siRNA on human hepatocellular carcinoma (HCC) cells, HepG2. The IC50 of ouabain (at 48 h) in HepG2 cells was 100 nmol/L, and this concentration was shown to induce cell cycle arrest as well as apoptosis. Interestingly, silencing of NKA α1 isoform could enhance the anti-cancer effect of ouabain, in between others, by increasing the expression of p21 Cip1 .
High EDLC plasma concentrations
(>100 nM-10 μM)
Effect on normal cells
Winnicka et al., [71] dealing with the dual role of cardenolides in human fibroblasts, demonstrated that ouabain, digoxin and proscillaridin A only at relatively high concentrations (>300 nM) increased intracellular Ca 2+ concentration, activated caspase-3 and induced apoptosis in human fibroblasts. Similarly, the group of Chueh et al. [72] revealed cytotoxic effects (apoptosis) of ouabain on human prostatic smooth muscle cells only at high concentrations. Akimova et al. [84] examined the role of MAPK in the death of ouabain-treated renal epithelial cells. Exposure of C7-MDCK cells to 3 μM ouabain led to phosphorylation of p38 and consequent apoptosis, without a significant impact on phosphorylation of ERK and JNK. In ouabain-resistant smooth muscle cells from rat aorta and endothelial cells from human umbilical vein, no effect of ouabain on p38 phosphorylation was observed.
Effect on malignant cells
Yeh et al. [85] demonstrated that digoxin, digitoxin and ouabain significantly inhibited the proliferation of human prostate cancer LNCaP, DU145 and PC3 cells at 1 μM or 10 μM. In contrast, normal control human glomerular epithelial cells showed no response to digitalis treatment at all tested doses. The discrepancy to the results of other authors (see above) remains to be clarified. Felth et al. [38] screened several cardiac glycosides in three human colon rectal cancer cell lines (HT29, HCT116 and CC20). Convallatoxin, oleandrin and proscillaridin A were identified as the most potent test compounds, with IC50 values ranging from 0.007 μM to 0.55 μM. Interestingly, the combination of digitoxin and oxaliplatin exhibited synergism in the otherwise highly drug-resistant HT29 cell line.
Comparison of NA/K-Atpase Characteristics in Tumor vs. Normal Cells
After summarizing these effects of DLC on different kinds of cell types in different experimental conditions, it becomes evident that the interaction between NKA and DLC follows a certain scheme according to the (E)DLC plasma concentrations [Figure 1]:
- At low EDLC plasma concentrations with no significant NKA inhibition, cell proliferation via Src/EGF-R/PI3K/Akt and Raf/Ras/MAPK pathways is predominant, resulting in transient ERK1/2 activation, activation of NF-kB and, in addition, recycling of signalosome members (α1, EGF-R).
- At low physiologic EDLC plasma concentrations with only partly (25%) NKA inhibition, cell cycle arrest and cell differentiation via the Src-EGFR-PI3K-Akt pathway are seen, resulting in sustained ERK1/2 activation with upregulation of p21 Cip and, in addition, lysosomal degradation of signalosome members.
- At high physiologic EDLC plasma concentrations, mainly “classic” NKA inhibition occurs, while pro-proliferative effects are diminished or blocked.
- At high EDLC plasma concentrations (with >60% NKA inhibition), cell apoptosis is revealed involving high intracellular Ca 2+ , production of ROS and activation of caspase-3.
This “stepwise” reaction scheme of NKA-EDLC interaction seems to be valid for both normal as well as tumor cells. What are the underlying mechanisms driving different cells either into cell growth, cell differentiation, cell cycle arrest or apoptosis when they are exposed to the same (E)DLC plasma concentrations? While dealing with this question, we will show that the “physiologic” EDLC range of normal cells indeed correlates with a specific response pattern that shifted to a lower “tumor-specific physiologic” range in tumor cells. According to Schoner, [45] “the mechanism by which a normal cell or a tumor cell enters the pathway of differentiation and proliferation or apoptosis seems to be essentially the same for normal and malignant cells.” Therefore, the crucial difference is likely to be found at the cell membrane surface. With respect to the DLC (ligands) and their specific receptor (NKA), it is well established that a distinct combination of α- and β-subunits is crucial for the kind of activated response. [86],[87],[88],[89] Not only have tissue-specific patterns of NKA isoforms [90],[91],[92] been identified but also a switch of isoforms during development from neonatal to adult tissues has been observed. [93],[94],[95] Crambert et al. [96] mention in a detailed analysis of the human NKA that the isozymes combined with the α1 isoform have the lowest Kd (i.e., the highest affinity for EDLC), the highest sensitivity toward K + and Na + and, hence, the greatest turnover, compared with other isozymes. These features reflect the “housekeeping role” of the ubiquitous α1 subunit, as “Na/K-ATPase with such characteristics should work at optimum rates under physiologic conditions but cannot respond to increased physiological demands.” [96] This implies that in case of transforming cells with, for example the “leaking-phenomenon” (see below), a “normal-active” NKA cannot respond to the needs of malignant cell transformation, like high intracellular K + – and glucose concentrations. Only after structural and/or conformational changes of the NKA (e.g., switch of isoforms) in the process of malignant transformation, can the pump provide this task, in between others, by developing hyperactivity.
Recent research has shed light on a new, still controversial role of α1 in cancer development and therapy. What seems to crystallize is the observation that in many different cancers (bladder, prostate, gastric), a reduced expression of the α1- and increased expression of the α3-isoform is present. [97],[98],[99],[101] The meaning of these switches in α-isoforms in cancer progression is not yet fully understood. On the one hand, the α1 subunit is essential for maintaining the actin cytoskeleton [102] and cell-growth capacity of a cell, most likely as part of the “signalosome” formed in special caveolae. [66],[67],[68] Kiss et al. called the α1 subunit a new target in cancer therapy, especially in NCSLC and Glioblastoma. [59],[60],[103] UNBS1450, a semi-synthetic cardenolide, decreased in NSCLC A549 cells both the NF-κB transcriptional activity and the DNA-binding capacity of the p65 subunit. [50] In Gioblastoma cells, the same compound caused ATP depletion, disorganization of the cytoskeleton and, finally, autophagic cell death. [103] Newman et al. [42] pointed out that, in addition to its growth-stimulatory effects, the α1 NKA isoform is associated with high intracellular gluthathione levels that prevent or delay ROS-induced apoptosis. This protective feature is “logic” for normal cells considering their genetic program, which drives them to proliferate unless they are aged, or dysfunctional (by mutation or other DNA damages). Xie et al. [49] demonstrated elegantly in a recent study that the knockout for NKA α1 is sufficient to upregulate the cell cycle inhibitor p21 Cip1 , leading to cell cycle arrest. Ouabain at low doses (nM) induced NKA endocytosis in both a benign and two malignant cell lines but, however, whereas in normal cells a repletion of NKA α1 at the cell membrane was seen (involving the Src/PI3K/Akt/mTOR pathway), which correlated with cell growth stimulation, in both malignant cell lines the NKA α1 subunits were directed to lysosomal degradation, resulting in cell growth inhibition. [49] The authors admit that the reason for this opposite behavior still needs to be clarified and suggested differences, e.g. in the components of the signalosome (caveolin-1 and cholesterol). We will propose below that mutations or amplifications of the EGF-R (as often found in malignancies) may be crucial determinants of α1 intracellular pathways and, hence, of the cell’s fate. In summary, for the sake of tumor growth control, the down-regulation or another form of inactivation of the NKA α1 isoform is “reasonable” and, thus, an important goal in targeted therapies.
On the other hand, as described above, many cancers (i.e. pancreatic) have already down-regulated NKA α1- and upregulated α3 isoforms. Contrary to what we might expect from the recently mentioned data, these cancers are not less aggressive than those with α1 expression. The reasons for this down-regulation of NKA α1 in some cancer types are not fully evaluated; most likely, cytokines are involved. From all available data, we may assume that endogenous DLC are “already-at-work.” In other words, one and the same cancer cell could have a different NKA isoform pattern during its life span with high expression of α1 isoforms in early stages and low α1 expression in later stages – in favor of high α3 – when endogenous defense mechanisms, including stress hormones like EDLC, have entered the “battlefield.” In any case, a high α3/α1 ratio seems to render cancer cells more sensitive toward cell growth inhibition and/or apoptosis. [42],[104] We may say in a literal sense that in cancer therapy, we continue or follow the path that nature itself has already chosen.
Concerning normal tissue, the NKA α3 subunit is predominant (α3/β1) in brain and neuronal tissue. As mentioned above, in cancer, the α3 isoform is often overexpressed. Shibuya et al. [100] analyzed the NKA isozyme expression in HCC. Interestingly, the expression levels of the NKA α3 isoform in HCC tissues were not only significantly higher than those in the accompanying non-tumor tissues but also correlated significantly with the NKA activity. The authors suggested that overexpression of α3 increases the Na/K-ATPase activity of HCC cells. [100] One possible interpretation is that in the program of malignant transforming cells, a signal for the need of more nutrition is translated in signaling cascades leading to a special NKA isoform pattern (α3/β2?) fulfilling these needs. But, indeed, the observed “correlation” between a3 expression and NKA activity could also represent a pure epiphenomenon. Remarkably, DLC not only have a high sensitivity to the human α3 subunit but also are able to downregulate the mRNA of the α3-subunit. [105] It is not yet clear whether the NKA α3 subunit per se in cancer cells has a specific tumor-promoting effect, but there is evidence that the upregulation of α3 is promoted by the pro-inflammatory surrounding that is typical for tumor formations. Besides, it has been demonstrated that, on the other hand, α3 downregulation is driving human leukemic cells into cell differentiation. [106] Remembering what we said above about the downregulation of α1 “in favor” of α3, you get the impression that the “devil is driven out by the Beelzebub.” Moreover, it has been demonstrated that the α3 isoform can substitute α1 in the signalosome and induce downstream signaling pathways. Pierre et al. [89] used the baculovirus expression system to determine which subunits of the transporter are required for mediating signal transduction events, e.g. the activation of ERK1/2 by phosphorylation. Interestingly, Sf9 insect cells expressing the NKA α1/β1 isozyme showed under ouabain application a dose-dependent linear increase in p-ERK, with the highest response obtained at 100 μM and 1000 μM. In contrast, Sf9 cells expressing the α3/β1 isozyme showed a dual response pattern, with increase in p-ERK1/2 only up to 0.1 μM and, afterwards, a decrease, reflecting an inhibitory effect of ouabain on cell growth-related pathways at higher concentrations. [89] This is in accordance with the known dual activity of DLC, and also supports our lower threshold theory in cancer (see below): the same ERK1/2-activating effect was obtained in α3/β1 (overexpressed in cancer) cells with 10 3 -10 4 -fold lower ouabain concentrations than in α1/β1 cells. The studies and results from baculovirus expression systems are limited in so far that they work with rodent NKA isoforms, with α1 having a much lower ouabain sensitivity than α2, α3 and α4. In human cells, in contrast, all isoforms are similarly sensitive to ouabain. [96] Assuming a comparable mechanism for the induction of signaling pathways, the data from rodents may be applied to human beings.
With respect to the NKA β subunits, it is accepted that β1-isoform expression in malignant cells is often downregulated, as shown for human clear cell renal, [107] gastric [108] and bladder cancer. [97] This β1-downregulation has been suggested to be associated with the loss of tight junctions and epithelial polarity in cancer cells. [109] It was also demonstrated that decreased expression of the β1-subunit in poorly differentiated carcinoma cell lines correlated with increased expression of the transcription factor Snail, known to downregulate E-cadherin, with consequent transition from epithelial to mesenchymal phenotypes. [110] Finally, Rajasekaran et al. [111] showed that the levels of phosphorylated ERK 1/2 are inversely correlated with the β1 isoform levels in the tumors (MSV-MDCK), indicating a direct tumor-suppressor function of β1 in epithelial cells. Blanco et al. [112] analyzed the function of different β isoforms, α3/β1 and α3/β2. Using Sf9 cells, they mentioned that the accompanying β subunit isoform does not drastically affect the properties of the α3 subunit. Both NKA isozymes have similar turnover numbers, affinities for K + and ATP and comparable high sensitivity to ouabain. Other authors claim that a switch from β1 to β2 may have an impact on tumorigenesis. [90] Here, further studies are needed, assuming that the downregulation of one isoform (e.g., β1) involves the upregulation of another one (e.g., β2). To summarize, in cancer, we often deal with overexpression of the α3 subunit and downregulation of the β1 subunit.
In malignant cells, not only the structure of the Na/K-ATPase is changed but also its dynamics. Decades ago, it was discovered that kinetic changes in the Na/K-ATPase activity are already present at very early stages of tumorigenesis, even long before their morphologic manifestation. [32],[113],[114],[115] Gonta-Grabriec et al. [116] revealed that in both spontaneous and radiation-induced thymomas, 86Rb uptake, ATP hydrolysis and 3H-ouabain binding per cell were higher than in normal thymuses. These changes correlated highly with cAMP content and 3H-thymidine incorporation, taken as indicators of the proliferative activity typical for a pre-leukemic period. Moreover, NKA activity may vary during the lifespan of malignant cells. For example, a depolarization of the plasma cell membrane in chicken embryo fibroblasts, transformed by Rous sarcoma virus, was described, reflecting a reduced NKA activity, maybe shortly before apoptosis. [117] Similarly, Davies et al. [118] measured the kinetics of the NKA in distal colonic mucosa of CF1 mice 1 week after injections of the carcinogen 1,2-dimethyhydrazine (DMH) over 4 weeks. The V max of the pump in pre-malignant mucosa was lower (55%-65% of control) for both sodium and potassium substrate activation, correlating with a 50% decreased NKA activity.
The reasons for these discrepancies are not yet clear. In malignant cell transformation, a “leakage phenomenon” was described causing a hyperactivity of the pump to compensate for the loss of potassium (Kaplan, 1978) and providing the tumor cell with nutrition necessary for the aberrant increased metabolism. [114] Whether and when exactly this hyperactive state is preceded (or followed) by a period of reduced pump activity is not known in detail. One possible scenario is that in a very early stage, the pump activity is reduced and only after, e.g. upregulation of the α3 isoform, the pump may switch to a hyperactive form (see: Shibuya). In general, basic kinetic properties of the NKA are modulated by a family of transmembrane spanning FXYD proteins that are colocalized with the NKA αβ subunits in the cell membrane. [119],[120] In tumorgenesis, they seem to be upregulated; for instance, in NSCLC, recently, a high overexpression of FXYD5 (related to ion channel) was revealed correlating with increased activity (V max ) of the pump, loss of TJ, increased cell permeability, impaired attachment and restricted cell movement. [120] Thus, the activity of the NKA might reflect the metabolic needs of a transforming cell, but the NKA that is embedded in the signalosome, is known to transmit signals independently of the pump’s activity.
Finally, analyzing the parameters at the cell membrane surface, we have to examine the neighborhood of the NKA. Considering that the NKA is closely interacting with other receptors at the cell surface (“signalosome”), it is reasonable to assume that a structural/functional change in one or more of these receptors will dramatically influence the kind of response to EDLC. This also helps to explain that malignant cells not only respond differently to DLC as compared with normal cells but also the differences in reaction between various cancer cell types.
The best analyzed receptor, EGF-R, is a tyrosinase kinase receptor, and its interaction with NKA in the signalosome is well established. [21],[24],[26] EGF-R kinase domain mutants found in non-small cell lung cancer (NSCLC) are constitutively active, a trait critical for malignant cell transformation. Chung et al. [121] stressed that aberrant trafficking of mutant EGF-R in NSCLC allows a preferential interaction with Src, a critical partner for EGF-R-mediated oncogenesis. Remarkably, mutant EGF-R, but not the wild-type EGF-R, show a perinuclear accumulation and colocalization with recycling endosomal markers such as Rab11 and EHD1 upon treatment of cells with endocytic recycling inhibitor monensin, suggesting that mutant EGF-Rs preferentially traffic through the endocytic recycling compartments. Medts et al. [122] aimed to test whether acute Src activation impacts on signalling and trafficking of non-liganded wild-type EGF-R. They found that thermoactivation caused rapid Src recruitment to the plasma membrane, concomitant association with EGF-R and its phosphorylation at Y845 and Tyr1173. Like low EGF concentrations, activated Src triggered EGF-R endocytosis via clathrin-coated vesicles and led to its sequestration in perinuclear/recycling endosomes with avoidance of multivesicular bodies and lysosomal degradation. The activation of Src and consecutive transactivation of (non-liganded) EGF-R by binding of ouabain to NKA is well documented. [21],[68],[123],[124] We want to add that endogenous EDLC may be the most important natural activators of Src and transactivators of EGF-R, competing with its primary ligand EGF. So far, some of these cited data would speak rather in favor of a tumor-promoting effect of EDLC by (a) contributing to enhanced recycling of the membrane-bound EGF-R and (b) by stimulating/activating the Src-EGFR-PI3K/Akt/mTor and Raf/Ras/MAPK pathways. How can we explain these discrepancies to the anti-proliferative effects described in numerous recent reports? We assumed previously that the mutation status of EGF-R might be a relevant cofactor in determining the direction of DLC-induced pathways. The above-mentioned aberrant trafficking of mutant EGF-R was observed in a setting where mutant EGF-R was stimulated by EGF, the natural ligand. [121] EGF binding induces auto-phosphorylation at Tyr1173 and Y845; ouabain, in contrast, has been shown to transactivate EGF-R by tyrosine phosphorylation at other sites. This could, at least partly, explain why the effects of DLC-transactivated EGF-R are different from EGF-stimulated EGF-R [Figure 2].
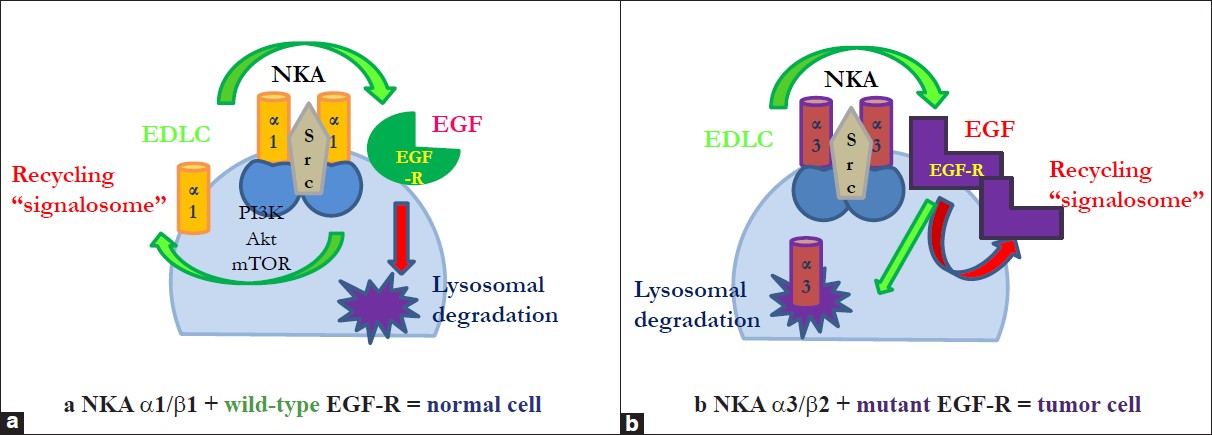 |
Figure 2: (a) Endogenous digitalis-like compounds (EDLC) and epidermal growth factor (EGF) induce different endocytotic trafficking pathways according to EGF-R mutation status and NKA isoforms. Transactivation of wt-epidermal growth factor receptor (EGF-R) by endogenous digitalis-like compounds induces endocytosis of the signalosome and causes recycling of the NKA α 1 subunit and other signalosome members at the cell membrane via activation of PI3K/Akt/mTor. Activation of wt-EGF-R by EGF, in contrast, results in lysosomal degradation of the signalosome, (b) Transactivation of mut-epidermal growth factor receptor (EGF-R) by endogenous digitalis-like compounds induces endocytosis of the signalosome and causes its lysosomal degradation. Activation of mut-EGF-R by EGF, in contrast, results in aberrant endocytic trafficking and recycling of EGF-R, NKA α 1/3 subunits and other signalosome members Click here to view |
Hypothesis
The facts presented above contribute to the hypothesis that the main reason for the observed phenomenon that tumor cells react differently to DLC as compared with normal cells is a lowered endogenous response threshold [Figure 1]. Above this threshold, tumor and normal cells reveal a similar response patterns toward (E)DLC, assuming no additional changes of intracellular molecules involved in the signaling cascades. In other words, tumor cells have their individual (tumor-specific) “physiologic EDLC ranges” starting at much lower (pmolar) EDLC concentrations as compared with normal cells:
Sub-threshold EDLC range of normal cells = “low-physiologic” EDLC range of tumor cells.
The reasons for a lowered threshold in malignant cells could be (as discussed above), i.e.
- Tumor-specific NKA pattern (α3/β2) at the cell membrane surface leading to changes in NKA sensitivity and activity
- Aberrations of receptors of the signalosome, e.g. mutant EGF-R
- Other changes in the signalosome, e.g. decrease of caveolin-1 and cholesterol
We assume that under physiologic conditions (normal serum K + and Na + concentrations), when the EDLC are binding preferentially to α3 isoforms (see: “K + -antagonism” Crambert), tumor cells with overexpression of α3 become, naturally, the selective target of EDLC while normal cells with predominant α1 isoforms are “protected.” This effect may be even more pronounced in situations of sub-physiologic low EDLC plasma concentrations, e.g. in chronic stress conditions with relative exhaustion of the adrenal gland and inability to maintain normal EDLC plasma concentrations. Very low (pmolar) EDLF plasma concentrations (pmolar) are critical in the following aspects:
- First, they will affect exclusively malignant cells because the pmolar range lies below the response threshold for normal cells.
- Secondly, they will stimulate exclusively their proliferation because this is the main function of DLC close to or above the response threshold [Figure 1].
- Finally, they might trigger or support the above outlined dynamic switch in NKA isoforms with loss of β1 (substituted by β2) and loss of α1 (substituted by α3), resulting in increased invasiveness and hyperactivity of the Na/K-ATPase.
To the best of our knowledge, there are no published data about the effect of pmolar DLC concentrations on malignant human cells. But, regarding the pro-proliferative effects of low DLC concentrations on non-malignant cells, one may expect a similar effect in the lowest “physiologic cancer” EDLC ranges. Here, we also want to mention the dual function of ERK1/2: transient activation leads to cell proliferation while sustained activation promotes cell cycle arrest and cell differentiation. [48] Low-dose ouabain has been shown to induce transient ERK1/2 activation, [28],[125],[126] whereas higher physiologic doses (≤100 nM) caused a sustained ERK1/2 activation with consequent upregulation of the cell cycle inhibitor p21 cip . [78] Applying this concept on our theory, we may draw the conclusion that both normal as well as tumor cells react with transient or sustained ERK1/2 activation according to their individual physiologic EDLC ranges [Figure 3].
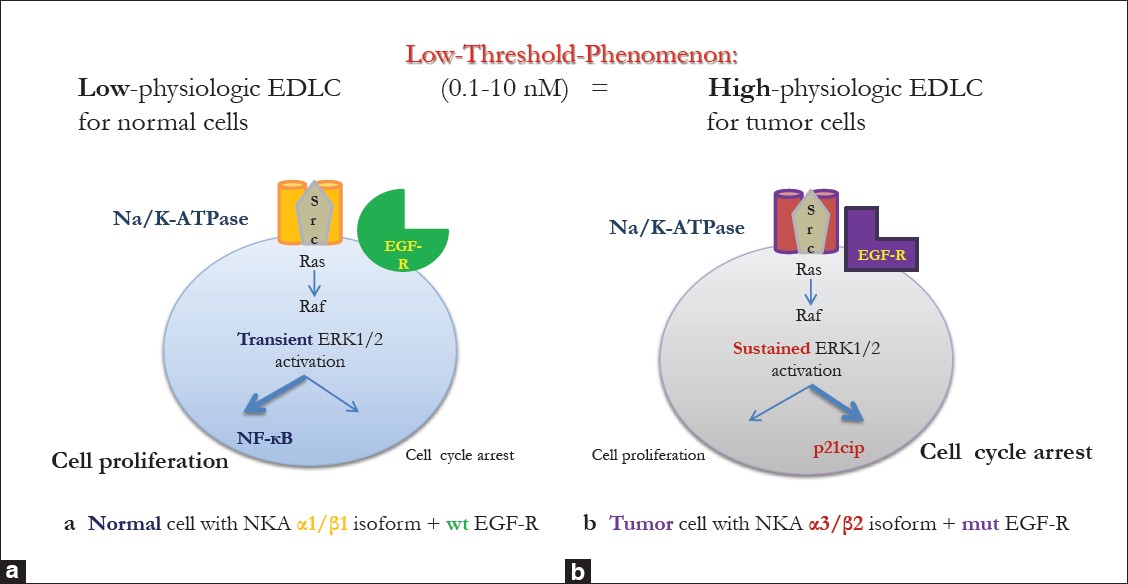 |
Figure 3: Induction of different signaling pathways in tumor vs. normal cells: impact of NKA isoforms and epidermal growth factor receptor mutation status. (a) In normal cells, low-physiologic endogenous digitalis-like compounds plasma concentrations (0.1 nM-10 nM) mainly stimulate cell proliferation, e.g. via transient activation of ERK 1/2 resulting in NF-kB induction, (b) For tumor cells, the same endogenous digitalis-like compounds concentrations represent a high-physiologic range and thus stimulate mainly cell cycle arrest via sustained activation of ERK 1/2 resulting in upregulation of Cip21 Click here to view |
Summary of Events in Early Tumorigenesis with Respect to the Role of NKA and EDLC
- Early changes of cell membrane composition and fluidity, induced either by biophysical factors of the environment (hypoxia, ROS), overexpression of oncogenes (Snail) or parts of the ion channels (FXYD), result in increased cell membrane permeability, downregulation of NKA β1, loss of tight junctions, aggravation of membrane permeability and loss of K +.
- Downregulation of NKA α1 and upregulation of NKA α3 by cytokines from an increasing pro-inflammatory tumor cell environment with compensatory increase in NKA activity result in restored intracellular K + and nutritional components (glucose, proteins). Other factors, e.g. overexpressed Bcl-2, may contribute to a hyperactivity of the NKA.
- High intracellular K + concentrations promote cell proliferation and also transformation by stimulating the expression of oncogenes (c-myc, c-fos).
- High intracellular glucose concentrations enhance excessive cell growth, especially when the tumor cell switches to aerobic glycolysis (Warburg effect).
- At this point – under normal conditions – upregulation of endogenous defense mechanisms, including the HPA system with the EDLC, help to re-balance the NKA isozymes and NKA amount at the cell surface membrane and trigger signal cascades leading to cell cycle arrest and apoptosis.
- In case of a deregulated HPA function (de-sensitization) with pathologic low EDLC supply from the adrenal gland, a breakdown of normal cell stability and integrity occurs, with further acquisition of malignant features (loss of adherence, increased invasiveness) and predominance of pro-proliferative effects via EDLC-NKA-Src-EGF-R interaction on tumor cells.
Outlook: Preliminary Results-Future Aims
Future clinical studies are necessary to verify this low threshold phenomenon and its tumor-promoting effect in situations of reduced EDLC plasma concentrations. A first step would be to analyze the effects of pmolar DLC concentrations on different cancer cell lines. A second step could include analyzing EDLC plasma concentrations of cancer patients at first diagnosis as compared with healthy persons, the NKA isozyme profile and NKA activity in the tumor tissue of these cancer patients correlated to known markers of tumor aggressiveness and the mutation status of the EGF-R. Furthermore, in order to verify the concept of a deregulated HPA axis (“De-sensitization”) as a cornerstone in tumorigenesis, it is warranted to evaluate, in between others, in the same patient and in control groups, the responses to acute stress (TSST) of both DLC and cortisol and to analyze their correlation.
We put forward the hypothesis that the ratio of these two adrenal stress hormones (DLC/cortisol) is prognostically relevant, rather than their absolute plasma/serum concentrations.
Here, we have to consider two controversial issues: first, the available data about physiologic EDLC plasma concentrations are limited and quite variable due to different methods and protocols [127],[128],[129],[130] [Table 1]. Second, the intracellular signaling cascades described in recent studies were often initiated by ouabain concentrations several orders of magnitude higher than the measured human plasma concentrations of putative endogenous ouabain. Hansen draws the conclusion that there is “No evidence for a role in signal-transduction of Na+/K+-ATPase interaction with putative endogenous ouabain.” [131] But, remembering that many of the studies used tissues from rodents with a known ouabain-insensitive α1-isoform of the NKA, we may assume that in human tissue, the signaling pathways altogether are triggered at lower DLC concentrations.
| Table 1: DLC and OLC plasma concentrations in human beings Click here to view |
In a pilot study, we aimed to analyze in healthy volunteers (n = 15) the plasma EDLC concentrations in correlation to cortisol (derived from saliva) by performing the mental stress test (TSST). For the first time, four specific responses (“cluster”) of EDLC to stress exposure were revealed [Figure 4]a-d. After establishing the EDLC cluster in healthy individuals, we analyzed the saliva cortisol concentrations corresponding to each of these EDLC clusters. We also discovered four distinct cortisol response patterns, but, interestingly, not always in positive correlation to EDLC [Figure 4]a-d. These results support our hypothesis that a dysbalance in EDLC/cortisol synthesis and secretion under prolonged stress exposure with inner “competition” may result in individually different risk patterns for cancer development (see: “EDLC cluster type 3”).
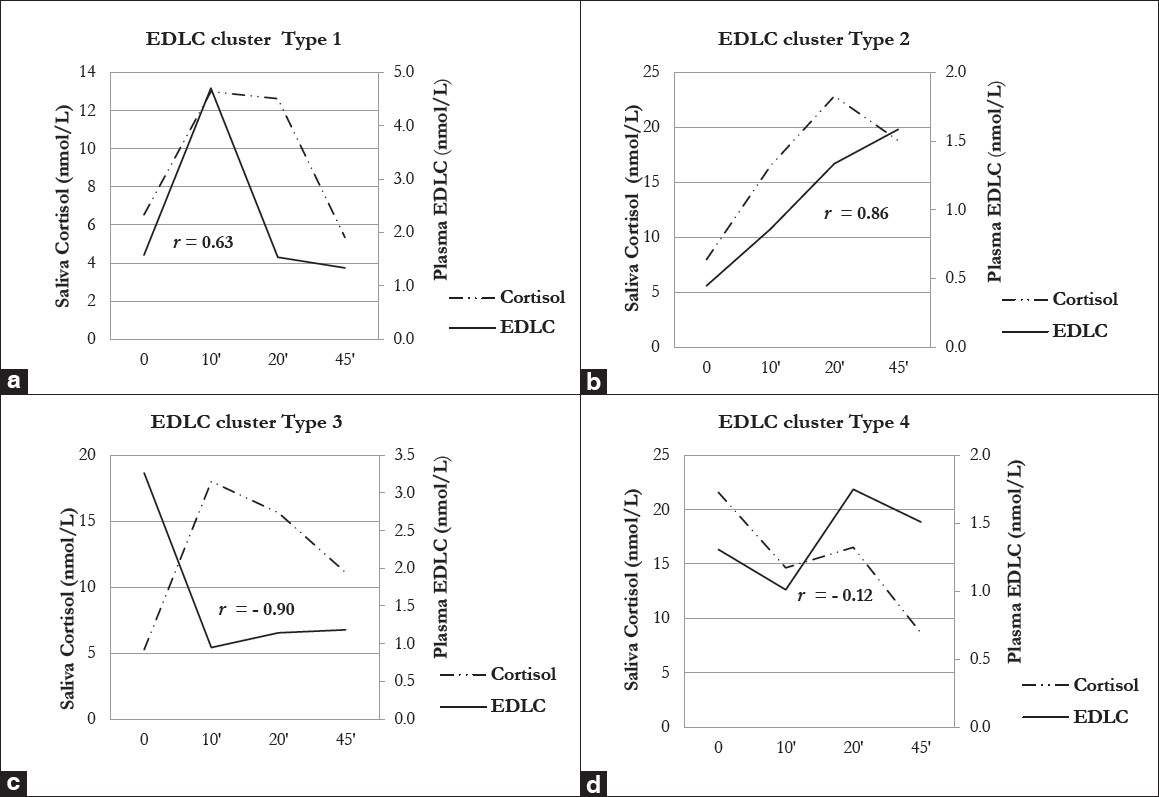 |
Figure 4: (a-d): Individual endogenous digitalis-like compounds “cluster” in response to mental stress (TSST) and their correlation to cortisol. (a) Endogenous digitalis-like compounds (EDLC) cluster Type 1 is characterized by normal baseline levels, a rapid and marked increase shortly (10′) after the stress test and a similar rapid and marked decrease afterwards. Cortisol in EDLC cluster Type 1 is characterized by normal baseline levels, a moderate increase shortly (10′) after the stress test, a plateau phase and a return to baseline at 45′. The correlation to EDLC is moderate (r = 0.63), but not significant, (b) Endogenous digitalis-like compounds (EDLC) cluster Type 2 is characterized by rather low baseline levels and a slow but continuous rise after the test (up to 45′). Cortisol in EDLC cluster Type 2 is characterized by normal baseline levels and a continuous increase starting shortly (10′) after the stress test, with a high peak at 20′. The correlation to EDLC is strong (r = 0.86), but not yet significant, (c) Endogenous digitalis-like compounds (EDLC) cluster Type 3 is characterized by very high baseline levels, a rapid and marked decrease shortly after the test (10′) and remaining low to normal levels. Cortisol in EDLC cluster Type 3 is characterized by normal baseline levels, a rapid and quite marked increase shortly (10′) after the test and a slow decrease afterwards. Interestingly, there is a strong inverse (r = – 0.90) and significant (P = 0.042) correlation to EDLC, (d) Endogenous digitalis-like compounds (EDLC) cluster Type 4 is characterized by low to normal baseline levels that do not rise after the test but remain all the time in the same range. Cortisol in EDLC cluster type 4 is characterized by extreme high baseline levels and a stepwise decrease starting shortly (10′) after the stress test. The correlation to EDLC is only weak (r = – 0.12) and not significant Click here to view |
In another preliminary trial (Registration ID NCT00310882), we analyzed EDLC plasma and cortisol serum concentrations in breast cancer patients (n = 22) at the time of first diagnosis compared with patients with a benign breast disease
(n = 10) as the control group. A significant positive correlation between EDLC and cortisol was seen in the control as well as in patients (r s = 0.7, P = 0.05), but only in cases of normal plasma/serum concentrations of both stress hormones [Figure 5]a. Interestingly, in breast cancer patients with very low EDLC plasma concentrations (<0.1 nmol/L), a significant inverse correlation (r s = – 0.9, P = 0.03) was observed [Figure 5]b. This is in accordance with our previous findings and supports our hypothesis that high “tumor-promoting” cortisol concentrations are maintained under chronic stress at the expense of “tumor-protecting” EDLC.
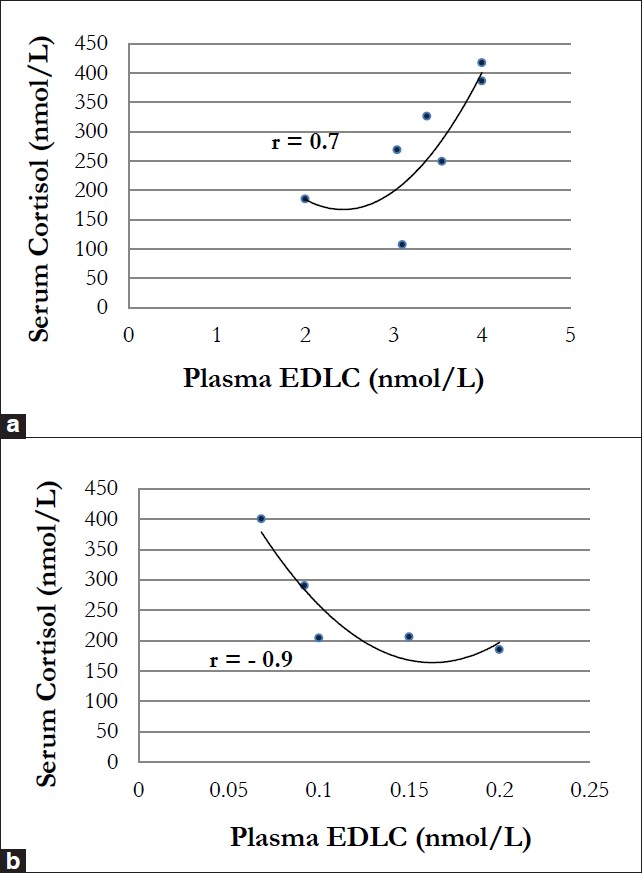 |
Figure 5: (a) Positive correlation between endogenous digitalis-like compounds (EDLC) and cortisol in patients with benign/ malignant breast disease. This correlation was only observed in patients with physiologic plasma EDLC (>0.1 nmol/L) and serum cortisol concentrations (r = 0.7, P = 0.05), (b) Inverse correlation between endogenous digitalis-like compounds (EDLC) and cortisol in patients with breast cancer. This negative correlation was observed only in cancer patients with subnormal (<0.1 nmol/L) plasma EDLC concentrations (r = -0.9, P = 0.03) Click here to view |
Conclusion
Assuming a lower threshold of malignant cells toward EDLC, it becomes evident that very low EDLC plasma concentrations due to an exhausted HPA system put an individual extremely at risk to develop cancer. It remains a challenging task to analyze in individuals their stress hormone response patterns and to investigate and develop tools to rebalance disturbed EDLC/cortisol concentrations – e.g., by physical (exercise) and mental (hypnosis, meditation) methods. This task should be started as early as possible in childhood to avoid the development of hormonal dysregulation and to strengthen the individual’s self-defense mechanisms against cancer.
Acknowledgment
The ideas and first results presented here developed during working in the Laboratory of David Lichtstein and the Oncologic Department of Tamar Peretz. For their continuous support, professionally and financially, the author would like to express great gratitude.
The author would also want to dedicate this paper to his deceased parents, Hannelore and Hartwig Weidemann, who encouraged him incessantly and supported the field of cancer research by a generous donation to the Hadassah Research Fund.
References
a
| 1. | Beck M, Szalay KS, Nagy GM, Tóth M, de Châtel R. Production of ouabain by rat adrenocortical cells. Endocr Res 1996;22:845-9.  |
| 2. | Hinson JP, Harwood S, Dawnay AB. Release of ouabain-like compound from the intact perfused rat adrenal gland. Endocr Res 1998;24:721-4. |
| 3. | Perrin A, Brasmes B, Chambaz EM, Defaye G. Bovine adrenocortical cells in culture synthesize an ouabain-like compound. Mol Cell Endocrinol 1997;126:7-15. |
| 4. | Doris PA. Immunological evidence that the adrenal gland is a source of an endogenous digitalis-like factor. Endocrinology 1988;123:2440-4. |
| 5. | Doris PA, Hayward-Lester A, Bourne D, Stocco DM. Ouabain production by cultured adrenal cells. Endocrinology 1996;137:533-9. |
| 6. | Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, et al. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci U S A 1991;88:6259-63. |
| 7. | Laredo J, Hamilton BP, Hamlyn JM. Ouabain is secreted by bovine adrenocortical cells. Endocrinology 1994;135:794-7. |
| 8. | Lichtstein D, Steinitz M, Gati I, Samuelov S, Deutsch J, Orly J. Biosynthesis of digitalis-like compounds in rat adrenal cells: Hydroxycholesterol as possible precursor. Life Sci 1998;62:2109-26. |
| 9. | el-Masri MA, Clark BJ, Qazzaz HM, Valdes R Jr. Human adrenal cells in culture produce both ouabain-like and dihydroouabain-like factors. Clin Chem 2002;48:1720-30. |
| 10. | Rauch AL, Buckalew VM Jr. Tissue distribution of an endogenous ligand to the Na, K ATPase molecule. Biochem Biophys Res Commun 1988;152:818-24. |
| 11. | Shah JR, Laredo J, Hamilton BP, Hamlyn JM. Different signaling pathways mediate stimulated secretions of endogenous ouabain and aldosterone from bovine adrenocortical cells. Hypertension 1998;31:463-8. |
| 12. | Sophocleous A, Elmatzoglou I, Souvatzoglou A. Circulating endogenous digitalis-like factor(s) in man is derived from the adrenals and its secretion is ACTH-dependent. J Endocrinol Invest 2003;26:668-74. |
| 13. | Weidemann H, Salomon N, Avnit-Sagi T, Weidenfeld J, Rosen H, Lichtstein D. Diverse effects of stress and additional adrenocorticotropic hormone on digitalis-like compounds in normal and nude mice. J Neuroendocrinol 2004;16:458-63. |
| 14. | Laredo J, Shah JR, Lu ZR, Hamilton BP, Hamlyn JM. Angiotensin II stimulates secretion of endogenous ouabain from bovine adrenocortical cells via angiotensin type 2 receptors. Hypertension 1997;29:401-7. |
| 15. | Muscella A, Greco S, Elia MG, Storelli C, Marsigliante S. Angiotensin II stimulation of Na+/K+ATPase activity and cell growth by calcium-independent pathway in MCF-7 breast cancer cells. J Endocrinol 2002;173:315-23. |
| 16. | Kaplan JH. Biochemistry of Na, K-ATPase. Annu Rev Biochem 2002;71:511-35. |
| 17. | Lingrel JB. Na, K-ATPase: isoform structure, function, and expression. J Bioenerg Biomembr 1992;24:263-70. |
| 18. | Mobasheri A, Avila J, Cózar-Castellano I, Brownleader MD, Trevan M, Francis MJ, et al. Na+, K+-ATPase isozyme diversity; comparative biochemistry and physiological implications of novel functional interactions. Biosci Rep 2000;20:51-91. |
| 19. | Scheiner-Bobis G. The sodium pump, its molecular properties and mechanics of ion transport. Eur J Biochem 2002;269:2424-33. |
| 20. | Aizman O, Aperia A. Na, K-ATPase as a signal transducer. Ann N Y Acad Sci 2003;986:489-96. |
| 21. | Li Z, Xie Z. The Na/K-ATPase/Src complex and cardiotonic steroid activated protein kinase cascades. Pflugers Arch 2009;457:635-44. |
| 22. | Caplan MJ. Ion pumps in epithelial cells: sorting, stabilization, and polarity. Am J Physiol 1997;272:G1304-13. |
| 23. | Contreras RG, Flores-Maldonado C, Lázaro A, Shoshani L, Flores-Benitez D, Larré I, et al. Ouabain binding to Na/K-ATPase relaxes cell attachment and sends a specific signal (NACos) to the nucleus. J Membr Biol 2004;198:147-58. |
| 24. | Haas M, Wang H, Tian J, Xie Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J Biol Chem 2002;277:18694-702. |
| 25. | Kometiani P, Li J, Gnudi L, Kahn BB, Askari A, Xie Z. Multiple signal transduction pathways link Na+/K+-ATPase to growth-related genes in cardiac myocytes. The roles of Ras and mitogen-activated protein kinases. J Biol Chem 1998;273:15249-56. |
| 26. | Liang M, Tian J, Liu L, Pierre S, Liu J, Shapiro JI, et al. Identification of a pool of non-pumping Na/K-ATPase. J Biol Chem 2007;282:10585-93. |
| 27. | Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. Ouabain interaction with cardiac Na, K-ATPase initiates signal cascades independent of changes in intracellular Na + and Ca 2+ concentrations. J Biol Chem 2000;275:27838-44. |
| 28. | Mohammadi K, Kometiani P, Xie Z, Askari A. Role of protein kinase C in the signal pathways that link Na, K-ATPase to ERK1/2. J Biol Chem 2001;276:42050-6. |
| 29. | Tian J, Xie ZJ. The Na, K-ATPase and calcium-signaling microdomains. Physiology (Bethesda) 2008; 23:205-11. |
| 30. | Xie Z, Cai T. Na+-K+–ATPase-mediated signal transduction: from protein interaction to cellular function. Mol Interv 2003;3:157-68. |
| 31. | Hartwell JL, Abbott BJ. Antineoplastic principles in plants: recent developments in the field. Adv Pharmacol 1969;7:117-209. |
| 32. | Kimelberg HK, Mayhew E. Increased ouabain-sensitive 86Rb+uptake and sodium and potassium ion-activated adenosine triphosphatase activity in transformed cell lines. J Biol Chem 1975;250:100-4. |
| 33. | Mayhew E, Levinson C. Reversibility of ouabain induced inhibition of cell division and cation transport in Ehrlich ascites cells. J Cell Physiol 1968;72:73-5. |
| 34. | Shiratori O. Growth inhibitory effect of cardiac glycosides and aglycones on neoplastic cells: in vitro and in vivo studies. Gann 1967;58:521-8. |
| 35. | Al-Ghoul M, Valdes R Jr. Mammalian cardenolides in cancer prevention and therapeutics. Ther Drug Monit 2008;30:234-8. |
| 36. | Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: Physiology, Pharmacology and novel therapeutic targets. Pharmacol Rev 2009;61:9-38. |
| 37. | Chen JQ, Contreras RG, Wang R, Fernandez SV, Shoshani L, Russo IH, et al. Sodium/potasium ATPase (Na, K-ATPase) and ouabain/related cardiac glycosides: a new paradigm for development of anti-breast cancer drugs? Breast Cancer Res Treat 2006;96:1-15. |
| 38. | Felth J, Rickardson L, Rosén J, Wickström M, Fryknäs M, Lindskog M, et al. Cytotoxic effects of cardiac glycosides in colon cancer cells, alone and in combination with standard chemotherapeutic drugs. J Nat Prod 2009;72:1969-4. |
| 39. | Haux J. Digitoxin is a potential anticancer agent for several types of cancer. Med Hypotheses 1999;53:543-8. |
| 40. | Johansson S, Lindholm P, Gullbo J, Larsson R, Bohlin L, Claeson P. Cytotoxicity of digitoxin and related cardiac glycosides in human tumor cells. Anticancer Drugs 2001;12:475-83. |
| 41. | Lopez-Lazaro M. Digitoxin as an anticancer agent with selectivity for cancer cells: possible mechanisms involved. Expert Opin Ther Targets 2007;11:1043-53. |
| 42. | Newman RA, Yang P, Pawlus AD, Block KI. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv 2008;8:36-49. |
| 43. | Prassas I, Diamandis EP. Novel therapeutic applications of cardiac glycosides. Nat Rev Drug Discov 2008;7:926-35. |
| 44. | Repke KR, Schön R, Megges R, Weiland J, Nissen E, Matthes E. Potential suitability of Na+/K(+)- transporting ATPase in pre-screens for anti-cancer agents. Anticancer Drug Des 1995;10:177-87. |
| 45. | Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism and cell growth. Am J Physiol Cell Physiol 2007;293:C509-36. |
| 46. | Winnicka K, Bielawski K, Bielawska A. Cardiac glycosides in cancer research and cancer therapy. Acta Pol Pharm 2006;63:109-15. |
| 47. | McConkey DJ, Lin Y, Nutt LK, Ozel HZ, Newman RA. Cardiac glycosides stimulate Ca 2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res 2000;60:3807-12. |
| 48. | Jing Y, Watabe M, Hashimoto S, Nakajo S, Nakaya K. Cell cycle arrest and protein kinase modulating effect of bufalin on human leukemia ML1 cells. Anticancer Res 1994;14:1193-8. |
| 49. | Tian J, Li X, Liang M, Liu L, Xie JX, Ye Q, et al. Changes in sodium pump expression dictate the effects of Ouabain on cell growth. J Biol Chem 2009;284:14921-9. |
| 50. | Mijatovic T, Op De Beeck A, Van Quaquebeke E, Dewelle J, Darro F, de Launoit Y, et al. The cardenolide UNBS1450 is able to deactivate nuclear factor KB-mediated cytoprotective effects in human non-small cell lung cancer cells. Mol Cancer Ther 2006;5:391-9. |
| 51. | Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci USA 2008;105:19579-86. |
| 52. | Bielawski K, Winnicka K, Bielawska A. Inhibition of DNA topoisomerases I and II, and growth inhibition of breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A. Biol Pharm Bull 2006;29:1493-7. |
| 53. | Mijatovic T, Mathieu V, Gaussin JF, De Nève N, Ribaucour F, Van Quaquebeke E, et al. Cardenolide-induced lysosomal membrane permeabilization demonstrates therapeutic benefits in experimental human non-small cell lung cancers. Neoplasia 2006;8:402-12. |
| 54. | Simpson CD, Mawji IA, Anyiwe K, Williams MA, Wang X, Venugopal AL, et al. Inhibition of the sodium potassium adenosine triphosphatase pump sensitizes cancer cells to anoikis and prevents distant tumor formation. Cancer Res 2009;69:2739-47. |
| 55. | Newman RA, Yang P, Hittelman WN, Lu T, Ho DH, Ni D, et al. Oleandrin-mediated oxidative stress in human melanoma cells. J Exp Ther Oncol 2006;5:167-81. |
| 56. | Newman RA, Kondo Y, Yokoyama T, Dixon S, Cartwright C, Chan D, et al. Autophagic cell death of human pancreatic tumor cells mediated by oleandrin, a lipid-soluble cardiac glycoside. Integr Cancer Ther 2007;6:354-64. |
| 57. | Pathak S, Multani AS, Narayan S, Kumar V, Newman RA. Anvirzel, an extract of Nerium oleander, induces cell death in human but not murine cancer cells. Anticancer Drugs 2000;11:455-63. |
| 58. | Sreenivasan Y, Raghavendra PB, Manna SK. Oleandrin-mediated expression of Fas potentiates apoptosis in tumor cells. J Clin Immunol 2006;26:308-22. |
| 59. | Mijatovic T, Roland I, Van Quaquebeke E, Nilsson B, Mathieu A, Van Vynckt F, et al. The alpha1 subunit of the sodium pump could represent a novel target to combat non-small cell lung cancers. J Pathol 2007;212:170-9. |
| 60. | Mijatovic T, Van Quaquebeke E, Delest B, Debeir O, Darro F, Kiss R. Cardiotonic steroids on the road to anti-cancer therapy. Biochim Biophys Acta 2007;1776:32-57. |
| 61. | Gaillard RC. Interaction between the hypothalamo-pituitary-adrenal axis and the immunological system. Ann Endocrinol (Paris) 2001;62:155-163. |
| 62. | Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory and preparative actions. Endocr Rev 2000;21:55-89. |
| 63. | Savino W, Dardenne M. Neuroendocrine control of thymus physiology. Endocr Rev 2000;21:412-43. |
| 64. | Tarcic N, Ovadia H, Weiss DW, Weidenfeld J. Restraint stress-induced thymic involution and cell apoptosis are dependent on endogenous glucocorticoids. J Neuroimmunol 1998;82:40-6. |
| 65. | Weidemann H. Na/K-ATPase, endogenous digitalis-like compounds and cancer development – a hypothesis. Front Biosci 2005;10:2165-76. |
| 66. | Cai T, Wang H, Chen Y, Liu L, Gunning WT, Quintas LE, et al. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J Cell Biol 2008;182:1153-69. |
| 67. | Liu L, Askari A. Beta-subunit of cardiac Na+-K+-ATPase dictates the concentration of the functional enzyme in caveolae. Am J Physiol Cell Physiol 2006;291:C569-78. |
| 68. | Xie Z. Ouabain interaction with cardiac Na/K-ATPase reveals that the enzyme can act as a pump and as a signal transducer. Cell Mol Biol (Noisy-le-grand) 2001;47:383-90. |
| 69. | Qiu J, Gao HQ, Zhou RH, Liang Y, Zhang XH, Wang XP, et al. Proteomics analysis of the proliferative effect of low-dose Ouabain on human endothelial cells. Biol Pharm Bull 2007;30:247-53. |
| 70. | Aydemir-Koksoy A, Allen JC. Low concentrations of Ouabain induce vascular smooth muscle cell proliferation. Cell Mol Biol (Noisy-le-grand) 2001;47:341-5. |
| 71. | Winnicka K, Bielawski K, Bielawska A, Miltyk W. Dual effects of Ouabain, Digoxin and Proscillaridin A on the regulation of apoptosis in human fibroblasts. Nat Prod Res 2010;24:274-85. |
| 72. | Chueh SC, Guh JH, Chen J, Lai MK, Teng CM. Dual effects of Ouabain on the regulation of proliferation and apoptosis in human prostatic smooth muscle cells. J Urol 2001;166:347-53. |
| 73. | Li J, Zelenin S, Aperia A, Aizman O. Low doses of ouabain protect from serum deprivation-triggered apoptosis and stimulate kidney cell proliferation via activation of NF-kappaB. J Am Soc Nephrol 2006;17:1848-57. |
| 74. | Khundmiri SJ, Metzler MA, Ameen M, Amin V, Rane MJ, Delamere NA. Ouabain induces cell proliferation through calcium-dependent phosphorylation of Akt (protein kinase B) in opossum kidney proximal tubule cells. Am J Physiol Cell Physiol 2006;291:C1247-57. |
| 75. | Wei Y, Xiao H, Jiang Y, Yang C, Zheng N. Low dose of Ouabain protects injury of spiral ganglion neurons in vitro. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2009;23:27-31. |
| 76. | de Rezende Corrêa G, Araujo dos Santos A, Frederico Leite Fontes C, Giestal de Araujo E. Ouabain induces an increase of retinal ganglion cell survival in vitro: the involvement of protein kinase C. Brain Res 2005;1049:89-94. |
| 77. | López-Lázaro M, Pastor N, Azrak SS, Ayuso MJ, Austin CA, Cortés F. Digitoxin inhibits the growth of cancer cell lines at concentrations commonly found in cardiac patients. J Nat Prod 2005;68:1642-5. |
| 78. | Kometiani P, Liu L, Askari A. Digitalis-induced signaling by Na+/K+-ATPase in human breast cancer cells. Mol Pharmacol 2005;67:929-36. |
| 79. | Winnicka K, Bielawski K, Bielawska A, Miltyk W. Apoptosis-mediated cytotoxicity of Ouabain, Digoxin and Proscillaridin A in the estrogen independent MDA-MB-231 breast cancer cells. Arch Pharm Res 2007;30:1216-24. |
| 80. | Huang YT, Chueh SC, Teng CM, Guh JH. Investigation of Ouabain-induced anticancer effect in human androgen-independent prostate cancer PC-3 cells. Biochem Pharmacol 2004;67:727-33. |
| 81. | Smith JA, Madden T, Vijjeswarapu M, Newman RA. Inhibition of export of fibroblast growth factor-2 (FGF-2) from the prostate cancer cell lines PC3 and DU145 by Anvirzel and its cardiac glycoside component, Oleandrin. Biochem Pharmacol 2001;62:469-72. |
| 82. | Li D, Qu X, Hou K, Zhang Y, Dong Q, Teng Y, et al. PI3K/Akt is involved in Bufalin-induced apoptosis in gastric cancer cells. Anticancer drugs 2009;20:59-64. |
| 83. | Xu ZW, Wang FM, Gao MJ, Chen XY, Hu WL, Xu RC. Targeting the Na(+)/K(+)-ATPase alpha1 subunit of hepatoma HepG2 cell line to induce apoptosis and cell cycle arresting. Biol Pharm Bull 2010;33:743-51. |
| 84. | Akimova OA, Lopina OD, Rubtsov AM, Gekle M, Tremblay J, Hamet P, et al. Death of ouabain-treated renal epithelial cells: evidence for p38 MAPK-mediated Na (i) (+) /K (i) (+) -independent signaling. Apoptosis 2009;14:1266-73. |
| 85. | Yeh JH, Huang WJ, Kan SF, Wang PS. Inhibitory effects of digitalis on the proliferation of androgen dependent and independent prostate cancer cells. J Urol 2001;166:1937-42. |
| 86. | Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol 1998;275:F633-50. |
| 87. | Jorgensen PL, Hakansson KO, Karlish SJ. Structure and mechanism of Na, K-ATPase: functional sites and their interactions. Annu Rev Physiol 2003;65:817-49. |
| 88. | Karpova L, Eva A, Kirch U, Boldyrev A, Scheiner-Bobis G. Sodium pump alpha1 and alpha3 subunit isoforms mediate distinct responses to Ouabain and are both essential for survival of human neuroblastoma. FEBS J 2010;277:1853-60. |
| 89. | Pierre SV, Sottejeau Y, Gourbeau JM, Sánchez G, Shidyak A, Blanco G. Isoform specificity of Na-K-ATPase-mediated Ouabain signaling. Am J Physiol Renal Physiol 2008;294:F859-66. |
| 90. | Akopyanz NS, Broude NE, Bekman EP, Marzen EO, Sverdlov ED. Tissue-specific expression of Na, K-ATPase beta-subunit. Does beta 2 expression correlate with tumorigenesis? FEBS Lett 1991;289:8-10. |
| 91. | Arystarkhova E, Sweadner KJ. Tissue-specific expression of the Na,K-ATPase beta3 subunit. The presence of beta3 in lung and liver addresses the problem of the missing subunit. J Biol Chem 1997;272:22405-8. |
| 92. | Huang L, Kometiani P, Xie Z. Differential regulation of Na/K-ATPase alpha-subunit isoform gene expressions in cardiac myocytes by ouabain and other hypertrophic stimuli. J Mol Cell Cardiol 1997;29:3157-67. |
| 93. | Corthésy-Theulaz I, Mérillat AM, Honegger P, Rossier BC. Differential regulation of Na/K-ATPase isoform gene expression by T3 during rat brain development. Am J Physiol 1991;261:C124-31. |
| 94. | Herrera VL, Cova T, Sassoon D, Ruiz-Opazo N. Developmental cell-specific regulation of Na(+)-K(+)-ATPase alpha 1-, alpha 2-, and alpha 3-isoform gene expression. Am J Physiol 1994;266:C1301-12. |
| 95. | Lopez LB, Quintas LE, Noël F. Influence of development on Na(+)/K(+)-ATPase expression: isoform- and tissue-dependency. Comp Biochem Physiol A Mol Integr Physiol 2002;131:323-33. |
| 96. | Crambert G, Hasler U, Beggah AT, Yu C, Modyanov NN, Horisberger JD, et al. Transport and pharmacological properties of nine different human Na, K-ATPase isozymes. J Biol Chem 2000;275:1976-86. |
| 97. | Espineda C, Seligson DB, James Ball W Jr, Rao J, Palotie A, Horvath S, et al. Analysis of the Na, K-ATPase alpha- and beta-subunit expression profiles of bladder cancer using tissue microarrays. Cancer 2003;97:1859-68. |
| 98. | Mobasheri A, Fox R, Evans I, Cullingham F, Martín-Vasallo P, Foster CS. Epithelial Na/K-ATPase is down-regulated in canine prostate cancer; a possible consequence of metabolic transformation in the process of prostate malignancy. Cancer Cell Int 2003;3:8. |
| 99. | Sakai H, Suzuki T, Maeda M, Takahashi Y, Horikawa N, Minamimura T, et al. Up-regulation of Na(+),K(+)-ATPase alpha 3-isoform and down-regulation of the alpha1-isoform in human colorectal cancer. FEBS Lett 2004;563:151-4. |
| 100. | Shibuya K, Fukuoka J, Fujii T, Shimoda E, Shimizu T, Sakai H, et al. Increase in ouabain-sensitive K+-ATPase activity in hepatocellular carcinoma by overexpression of Na+, K+-ATPase alpha 3-isoform. Eur J Pharmacol 2010;638:42-6. |
| 101. | Yang P, Menter DG, Cartwright C, Chan D, Dixon S, Suraokar M, et al. Oleandrin-mediated inhibition of human tumor cell proliferation: Importance of Na, K-ATPase α subunits as drug targets. Mol Cancer Ther 2009;8:2319-28. |
| 102. | Nelson WJ, Hammerton RW. A membrane-cytoskeletal complex containing Na+,K+-ATPase, ankyrin, and fodrin in Madin-Darby canine kidney cells: implications for the biogenesis of epithelial cell polarity. J Cell Biol 1989;108:893-902. |
| 103. | Lefranc F, Kiss R. The sodium pump alpha1 subunit as a potential target to combat apoptosis-resistant Glioblastomas. Neoplasia 2008;10:198-206. |
| 104. | Lin Y, Ho DH, Newman RA. Human tumor cell sensitivity to oleandrin is dependent on relative expression of Na+, K+ -ATPase subunitst. J Exp Ther Oncol 2010;8:271-86. |
| 105. | Kometiani P, Tian J, Li J, Nabih Z, Gick G, Xie Z. Regulation of Na/K-ATPase beta 1-subunit gene expression by Ouabain and other hypertrophic stimuli in neonatal rat cardiac myocytes. Mol Cell Biochem 2000;215:65-72. |
| 106. | Gilmore-Hebert M, Schneider JW, Greene AL, Berliner N, Stolle CA, Lomax K, et al. Expression of multiple Na+,K+-adenosine triphosphatase isoform genes in human hematopoietic cells. Behavior of the novel A3 isoform during induced maturation of HL60 cells. J Clin Invest 1989;84:347-51. |
| 107. | Rajasekaran SA, Ball WJ Jr, Bander NH, Liu H, Pardee JD, Rajasekaran AK. Reduced expression of beta-subunit of Na/K-ATPase in human clear cell renal cell carcinoma. J Urol 1999;162:574-80. |
| 108. | Avila J, Lecuona E, Morales M, Soriano A, Alonso T, Martín-Vasallo P. Opposite expression pattern of the human Na, K-ATPase beta 1 isoform in stomach and colon adenocarcinomas. Ann N Y Acad Sci 1997;834:653-5. |
| 109. | Rajasekaran AK, Rajasekaran SA. Role of Na-K-ATPase in the assembly of tight junctions. Am J Physiol Renal Physiol 2003;285:F388-96. |
| 110. | Espineda CE, Chang JH, Twiss J, Rajasekaran SA, Rajasekaran AK. Repression of Na/K-ATPase beta1-subunit by the transcription factor Snail in carcinoma. Mol Biol Cell 2004;15:1364-73. |
| 111. | Inge LJ, Rajasekaran SA, Yoshimoto K, Mischel PS, McBride W, Landaw E, et al. Evidence for a potential tumor suppressor role for the Na/K-ATPase ß1-subunit. Histol Histopathol 2008;23:459-67. |
| 112. | Blanco G, Sánchez G, Mercer RW. Comparison of the enzymatic properties of the Na,K-ATPase alpha 3 beta 1 and alpha 3 beta 2 isozymes. Biochemistry 1995;34:9897-903. |
| 113. | Shen SS, Hamamoto ST, Bern HA, Steinhardt RA. Alteration of sodium transport in mouse mammary epithelium associated with neoplastic transformation. Cancer Res 1978;38:1356-61. |
| 114. | Kaplan JG. Membrane cation transport and the control of proliferation of mammalian cells. Annu Rev Physiol 1978;40:19-41. |
| 115. | Latzkovits L, Torday C, Jánossy T, Erdös E. Manifestation of K+ transport alterations in cultered tumour cells of mice. Acta Chir Hung 1983;24:287-94. |
| 116. | Gonta-Grabiec K, Rossowski W, Szumiel I. Properties of Na/K-ATPase and alkaline phosphatase alter during spontaneous and radiation-induced leukemogenesis in mice. Neoplasma 1986;33:141-55. |
| 117. | Banerjee SP, Bosmann HB, Morgan HR. Oncogenic transformation of chick-embryo fibroblasts by Rous sarcoma virus alters rubidium uptake and Ouabain binding. Exp Cell Res 1977;104:111-7. |
| 118. | Davies RJ, Sandle GI, Thompson SM. Inhibition of the Na, K-ATPase pump during induction of experimental colon cancer. Cancer Biochem Biophys 1991;12:81-94. |
| 119. | Geering K. FXYD proteins: new regulators of Na-K-ATPase. Am J Physiol Renal Physiol 2006;290:F241-50. |
| 120. | Lubarski I, Pihakaski-Maunsbach K, Karlish SJ, Maunsbach AB, Garty H. Interaction with the Na, K-ATPase and tissue distribution of FXYD5 (related to ion channel). J Biol Chem 2005;280:37717-24. |
| 121. | Chung BM, Raja SM, Clubb RJ, Tu C, George M, Band V, et al. Aberrant trafficking of NSCLC-associated EGFR mutants through the endocytic recycling pathway promotes interaction with Src. BMC Cell Biol 2009;10:84. |
| 122. | Medts T, de Diesbach MT, Cominelli A, N’Kuli F, Tyteca D, Courtoy PJ. Acute ligand-independent Src activation mimics low EGF-induced EGFR surface signaling and redistribution into recycling endosomes. Exp Cell Res 2010;316:3239-53. |
| 123. | Donepudi M, Resh MD. c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling. Cell Signal 2008;20:1359-67. |
| 124. | Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem 2000;275:27832-7. PubMed PMID:10874030. |
| 125. | Jin RM, Bai Y, Xiong AX, Lin W, Yu H, Wu XY, et al. MAPK signal pathway plays a role in proliferation of Jurkat cells induced by Ouabain. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2005;13:126-9. |
| 126. | Mohammadi K, Liu L, Tian J, Kometiani P, Xie Z, Askari A. Positive inotropic effect of Ouabain on isolated heart is accompanied by activation of signal pathways that link Na+/K+-ATPase to ERK1/2. J Cardiovasc Pharmacol 2003;41:609-14. |
| 127. | Bauer N, Müller-Ehmsen J, Krämer U, Hambarchian N, Zobel C, Schwinger RH, et al. Ouabain-Like compound changes rapidly on physical exercise in humans and dogs effects of beta-Blockade and Angiotensin-converting enzyme inhibition. Hypertension 2005;45:1024-8. |
| 128. | Berendes E, Cullen P, Van Aken H, Zidek W, Erren M, Hübschen M, et al. Endogenous glycosides in critically ill patients. Crit Care Med 2003;31:1331-7. |
| 129. | Lin MH, Liao CP, Lee JS, Chin YW, Hsu CC, Wei JS. Detection of endogenous digitalis-like immunoreactive factors in human blood. Proc Natl Sci Counc Repub China B 1998;22:129-35. PubMed PMID:9779602. |
| 130. | Li J, Wu Z, Xu J, Zhou Y, Chu X. Determination of digoxin-like immunoreactive substances in sera of 15 elder patients with cardiac insufficiency. Yao Xue Xue Bao 1998;33:655-8. |
| 131. | Hansen O. No evidence for a role in signal-transduction of Na+/K+-ATPase interaction with putative endogenous ouabain. Eur J Biochem 2003;270:1916-9. |
Authors
Dr. Heidrun Weidemann, Heidrun L.C. Weidemann, Sharett Institute of Oncology, Hadassah-Hebrew University, Medical Center, Jerusalem, Israel
Figures
[Figure 1], [Figure 2], [Figure 3], [Figure 4], [Figure 5]
Tables
[Table 1]