Masahito Shimizu, Yohei Shirakami, Kenji Imai, Koji Takai, Hisataka Moriwaki
Department of Medicine, Gifu University Graduate School of Medicine 1-1 Yanagido, Gifu 501-1194, Japan
| Date of Submission | 22-Feb-2012 |
| Date of Acceptance | 15-Jul-2012 |
| Date of Web Publication | 30-Aug-2012 |
Correspondence Address:
Masahito Shimizu
Department of Medicine, Gifu University Graduate School of Medicine 1-1 Yanagido, Gifu 501-1194
Japan
Source of Support: This work was supported in part by Grantsin-Aid from the Ministry of Education, Science, Sports, and Culture of Japan (No. 22790638 to M. S. and No. 21590838 to H. M.) and by a Grant-in-Aid for the 3rd Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan, Conflict of Interest: None
DOI: 10.4103/1477-3163.100398
Abstract
One of the key features of hepatocellular carcinoma (HCC) is the high rate of intrahepatic recurrence that correlates with poor prognosis. Therefore, in order to improve the clinical outcome for patients with HCC, development of a chemopreventive agent that can decrease or delay the incidence of recurrence is a critical issue for urgent investigation. Acyclic retinoid (ACR), a synthetic retinoid, successfully improves HCC patient survival by preventing recurrence and the formation of secondary tumors. A malfunction of the retinoid X receptor-α (RXRα) due to phosphorylation by the Ras-MAPK signaling pathway plays a critical role in liver carcinogenesis, and ACR exerts chemopreventive effects on HCC development by inhibiting RXRα phosphorylation. Here, we review the relationship between retinoid signaling abnormalities and liver disease, the mechanisms of how RXRα phosphorylation contributes to liver carcinogenesis, and the detailed effects of ACR on preventing HCC development, especially based on the results of our basic and clinical research. We also outline the concept of “clonal deletion and inhibition” therapy, which is defined as the removal and inhibition of latent malignant clones from the liver before they expand into clinically detectable HCC, because ACR prevents the development of HCC by implementing this concept. Looking toward the future, we discuss “combination chemoprevention” using ACR as a key drug since it can generate a synergistic effect, and may thus be an effective new strategy for the prevention of HCC.
Keywords: Acyclic retinoid, chemoprevention, clonal deletion and inhibition, combination therapy, HCC, RXRα phosphorylation
How to cite this article:
Shimizu M, Shirakami Y, Imai K, Takai K, Moriwaki H. Acyclic retinoid in chemoprevention of hepatocellular carcinoma: Targeting phosphorylated retinoid X receptor-α for prevention of liver carcinogenesis. J Carcinog 2012;11:11
How to cite this URL:
Shimizu M, Shirakami Y, Imai K, Takai K, Moriwaki H. Acyclic retinoid in chemoprevention of hepatocellular carcinoma: Targeting phosphorylated retinoid X receptor-α for prevention of liver carcinogenesis. J Carcinog [serial online] 2012 [cited 2021 Oct 14];11:11. Available from: https://carcinogenesis.com/text.asp?2012/11/1/11/100398
Introduction
Hepatocellular carcinoma (HCC) is one of the most frequently occurring cancers in the world. The number of new cases per year is estimated to be 750,000; approximately the same number (700,000) of people die from this malignancy each year. [1],[2] Chronic inflammation and subsequent cirrhosis of the liver, most cases of which are induced by persistent infection with hepatitis B virus (HBV) or hepatitis C virus (HCV), contribute to the development of HCC. [2],[3] In order to improve the prognosis of HCC, several effective strategies to prevent the development of primary HCC and intrahepatic recurrence of this malignancy have been demonstrated in clinical trials. A meta-analysis reported that antiviral treatment reduces the risk of HBV-related HCC recurrence and decreases liver-related mortality as well as overall mortality. [4] For patients with HCV-related HCC, the effectiveness of interferon therapy for preventing the recurrence of HCC has been shown by meta-analyses. [5],[6]
In addition to studies of antiviral treatment, other important trials using specific agents have been conducted to seek ways for preventing HCC development. For instance, supplementation with branched-chain amino acids reduces the risk of HCC in cirrhotic patients who are obese. [7] We reported the results of our prospective randomized study, [8],[9],[10] in which the oral administration of acyclic retinoid (ACR), a synthetic retinoid, inhibited the development of a second primary HCC and thus improved patient survival. The pleiotropic effects of ACR in the prevention of HCC and suppression of cancer cell growth have also been revealed in many experimental studies. [11],[12],[13] The aim of this article is to review the detailed effects of ACR in preventing the development of HCC, based on our clinical and basic research. In particular, we focus on the effects of ACR in targeting the phosphorylation of retinoid X receptor-α (RXRα), because malfunction of this nuclear receptor due to aberrant phosphorylation is closely involved in liver carcinogenesis. [14] We also review the concept of “clonal deletion and inhibition” therapy, a practical approach to preventing HCC development. Finally, we discuss the possibility of “combination chemoprevention,” which uses ACR as a key drug and is expected to be an effective strategy that takes advantage of pharmacologic synergism to prevent the formation of HCC.
Retinoids and their receptors
Retinoids, a group of structural and functional derivatives of vitamin A, have fundamental effects on cellular activities, including growth, differentiation, and apoptosis, as well as on morphology. [15],[16],[17] Retinoids exert their biologic functions primarily by regulating gene expression through two distinct nuclear receptors, the retinoic acid receptors (RARs) and RXRs, which are ligand-dependent transcription factors. Both RARs and RXRs are composed of three subtypes (α, β, and γ), which are characterized by a modular domain structure. [15],[16],[17] RARs can be activated by both all-trans-retinoic acid and 9-cis-retinoic acid with similar affinities, whereas RXRs are exclusively activated by 9-cis-retinoic acid.
After ligand binding, RXRs form homodimers as well as heterodimers with RARs, which interact with the retinoid X response element (RXRE) or the retinoic acid receptor responsive element (RARE), located in the promoter region of target genes, thereby modulating gene expression. [15],[16],[17] In addition to forming a heterodimer with RARs, RXRs can also form heterodimers with several other nuclear receptors, indicating that RXRs act as common heterodimerization partners for various types of nuclear receptors. [16] For instance, RXRs interact with peroxisome proliferator-activated receptors (PPARs), which are receptors for fatty acids, thus regulating PPAR-mediated pathways and controlling energy homeostasis. [18] Therefore, RXRs function as auxiliary factors, determining the effects of other hormones, and acting as master regulators of nuclear receptors. [16]
Retinoid abnormalities and liver disease
The liver is one of the most important target organs for retinoid actions. Hepatocytes play central roles in the uptake and processing of dietary retinol in the liver and secrete retinol-binding protein into the plasma. Hepatic stellate cells (HSCs) are critically involved in the storage of retinoids in the liver, suggesting that the development of hepatic disease is highly correlated with impaired hepatic retinoid metabolism and storage. [19],[20] In alcoholic patients, diminished hepatic retinoid storage is associated with progressively worsening stages of hepatic disease. [21] The loss of hepatic retinyl ester stores from the lipid droplets within HSCs leads to HSC activation and the development of liver fibrosis. [22],[23] A progressive decrease in serum retinol levels has also been observed in patients diagnosed with liver cirrhosis compared with healthy subjects, and those patients with both cirrhosis and HCC had significantly lower levels than patients with cirrhosis alone. [24],[25]
Several experimental studies using genetically engineered mice have revealed the pivotal effects of retinoids on fat metabolism in the liver. Yanagitani et al.[26] reported that RARα dominant-negative form transgenic mice developed steatohepatitis through the downregulation of hepatic mitochondrial β-oxidation activity of fatty acids. Studies in hepatocyte RXRα-deficient mice also demonstrated that RXRα plays vital roles in fatty acid and cholesterol metabolism in the liver. [27],[28] These observations suggest that retinoids and their receptors are involved in the mediation of normal hepatic lipid metabolism.
Retinoid abnormalities and liver carcinogenesis
As previously noted, retinoids and their receptors, especially RXRs, play an essential role in controlling normal cell proliferation, differentiation, metabolism, and death (regulation of apoptosis). [15],[16],[17] On the other hand, these facts also suggest that the loss of retinoid activity or responsiveness is linked to deviation from normal cell proliferation and death, which are key factors for cancer development. [29],[30] Indeed, it is well established that abnormalities in retinoid signaling are prominently involved in carcinogenesis in several organs, including the liver. In a rodent model of liver carcinogenesis, [31] retinol was observed to be locally deficient in HCC lesions, but not in the adjacent normal liver tissues. Functional loss of retinoic acid leads to the occurrence of cellular dysplasia and cancer in the liver. [26] Overexpression of cellular retinoic acid-binding protein-II, which shows a high affinity for all-trans-retinoic acid, is associated with induction of retinoic acid resistance in HCC cells. [32] On the other hand, lecithin:retinol acyltransferase knockout mice, which possess increased retinoid signaling in the liver, are less susceptible to diethylnitrosamine (DEN)-induced hepatocarcinogenesis. [33] These findings suggest that supplementation with additional retinoids and improvement of retinoid signaling may be a promising strategy for the prevention and/or treatment of HCC.
In addition to retinoid depression, abnormalities in retinoid receptors are also associated with liver carcinogenesis and the growth of HCC cells. The RAR β gene, a tumor suppressor gene, is an HBV integration site, and the expression of this gene is markedly decreased in human HCC. [34],[35] RARβ expression is also suppressed in liver cancer cell lines. [36],[37] We have previously reported that the levels of RARβ protein and mRNA were decreased in HCC lesions in a rat model of chemically induced liver carcinogenesis. [38] On the other hand, RARγ is overexpressed in HCC tissues and cells, which is associated with the growth of HCC cells. [39] It has been reported that RARγ often resides in the cytoplasm of HCC cells and interacts with the p85α regulatory subunit of phosphatidylinositol 3-kinase, resulting in the activation of Akt and nuclear factor-κB, which are critical regulators of the growth and survival of cancer cells. [39],[40] These reports support an oncogenic potential for RARγ in liver carcinogenesis.
RXRα phosphorylation and HCC
Among retinoid receptors, RXRα is most abundant in the liver; therefore, its alterations are particularly implicated in the development and progression of HCC. RXRα is reported to bind to the enhancer element of HBV and modulate viral replication. [41] The expression of RXRα is decreased not only in HCC and liver cell adenoma, but also in glutathione S-transferase placental form-positive foci, a precancerous HCC lesion, as seen in a rat model of chemically induced liver carcinogenesis. [38] These findings indicate that repression of RXRα occurs even in the early stage of liver carcinogenesis.
Moreover, we have shown that aberrant phosphorylation of RXRα is critically involved in liver carcinogenesis. Initially, we revealed that the RXRα protein is anomalously phosphorylated at the serine and threonine residues and that it was seen to accumulate in both surgically resected human HCC tissues and HCC cell lines, whereas in normal hepatocytes, RXRα is unphosphorylated and is broken into smaller peptides. [14],[42] We previously reported that the phosphorylated form of RXRα protein was present in higher concentrations in HCC tissues than in noncancerous surrounding and normal liver tissues among all 10 cases examined. [14] Activated extracellular signal-regulated kinase (ERK) is highly expressed in HCC cells, and constitutive phosphorylation at the serine at position 260 of RXRα, a mitogen-activated protein kinase (MAPK)-ERK consensus site, is closely associated with its retarded degradation, low transcriptional activity, and promotion of cancer cell growth. In turn, the abrogation of phosphorylation by an MAPK inhibitor or transfection with unphosphomimic mutant RXRα restores the degradation of RXRα in a ligand-dependent manner. [14],[43] Phosphorylated RXRα abolishes its ability to form heterodimers with RARβ, and this is associated with uncontrolled cell growth and resistance to retinoids. [44] Moreover, phosphorylated RXRα is resistant to proteolytic degradation via the ubiquitination/proteasome-mediated pathway in human HCC cells, resulting in an accumulation of this phosphorylated protein within the HCC tissues. [45] Therefore, in HCC tissues and cells, the accumulation of phosphorylated RXRα, which is regarded as the nonfunctional form of RXRα, may interfere with the function of normal (unphosphorylated) RXRα in a dominant-negative manner. Our observations suggest that not only retinoid depletion, but also malfunction of retinoid receptors, especially phosphorylation of RXRα, may play a critical role in HCC development [Figure 1].
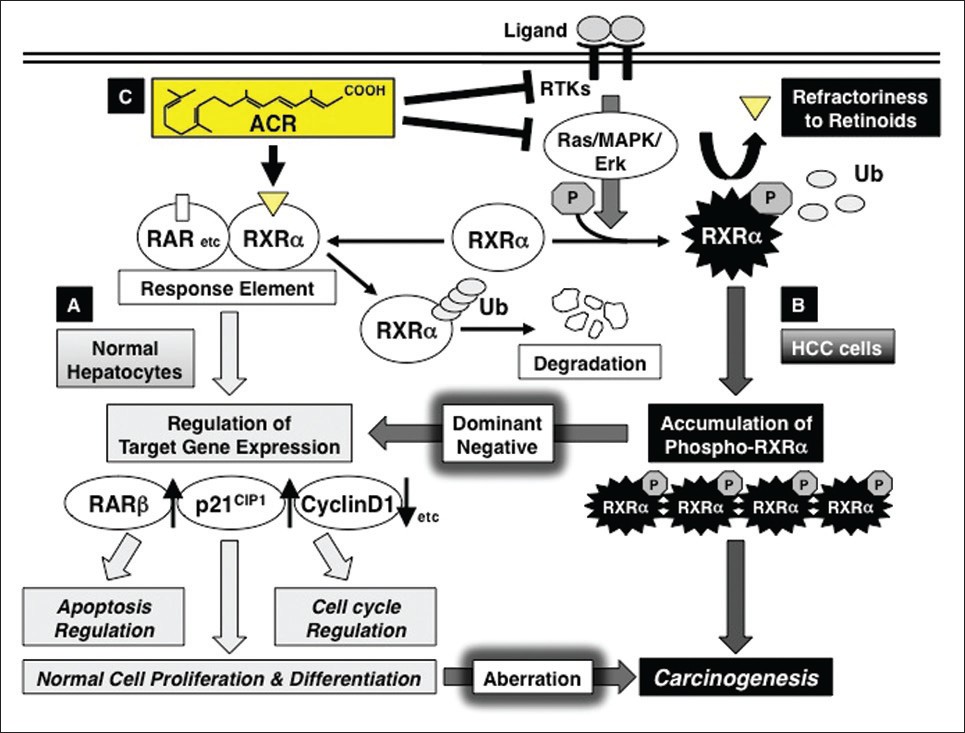 |
Figure 1: Retinoid refractoriness due to phosphorylation of retinoid X receptor alpha (RXR α), and its restoration by acyclic retinoid (ACR) in liver carcinogenesis. (a) In normal hepatocytes, when ACR binds to and activates RXR α, it forms homo- and/or heterodimers with other nuclear receptors, including retinoic acid receptors (RARs). This results in expression of the target genes, such as RAR β, p21 CIP1 , and cyclin D1, which regulate normal cell proliferation and differentiation, as well as controlling the induction of apoptosis and cell cycle progression. Thereafter, RXR α is rapidly ubiquitinated (Ub) and degraded via the proteasome pathway. (b) In hepatocellular carcinoma (HCC) cells, the Ras-mitogen-activated protein kinase (MAPK) pathway is highly activated and phosphorylates RXR α at serine residues, impairing dimer formation and the subsequent transactivation functions of the receptor (refractoriness to retinoids). Furthermore, nonfunctional phosphorylated RXR α is sequestered from ubiquitin/proteasome-mediated degradation and accumulates in liver cells. This interferes with the physiologic function of the remaining unphosphorylated (ie, functional) RXR α in a dominant-negative manner, causing a deviation from normal cell proliferation and differentiation, thereby playing a critical role in liver carcinogenesis. (c) ACR is not only a ligand for RXR α, but also a suppressor of the Ras-MAPK signaling pathway; it inhibits RXR α phosphorylation, thereby restoring the function of the receptor and activating the transcriptional activity of the responsive element. ACR also inhibits, directly or indirectly, the ligand (growth factor)-dependent RTK activities, which contribute to the inhibition of ERK and RXR α phosphorylation and suppression of growth in HCC cells Click here to view |
Mechanisms of ACR in HCC chemoprevention
ACR, which is the same substance as NIK-333 and Peretinoin (Kowa Pharmaceutical Co., Tokyo, Japan), inhibits both chemically induced liver carcinogenesis in rats and mice and spontaneously occurring HCC in mice. [31],[46],[47],[48],[49] ACR was initially developed as an agonist for both RXR and RAR. [50],[51] Therefore, this agent activates the promoter activity of RXRE and RARE and controls the expression of target genes, including RARβ, p21 CIP1 , and cyclin D1, which results in induction of apoptosis, cell cycle arrest in G 0 -G 1 , and growth inhibition in human HCC-derived cells. [43],[52],[53],[54],[55],[56],[57],[58],[59],[60],[61],[62] These findings suggest that ACR suppresses HCC, at least in part, by working as a ligand for retinoid receptors and controlling their target genes, especially RARβ and p21 CIP1 .
On the other hand, many experimental studies have shown that ACR exerts chemopreventive effects in HCC cells by inhibiting RXRα phosphorylation. In human HCC cells, ACR restores RXRα function by inactivating the Ras-MAPK signaling system, leading to dephosphorylation of RXRα. [43] ACR also suppresses cancer cell growth by inhibiting the activation and expression of several types of growth factors and their corresponding receptor tyrosine kinases (RTKs). [47],[48],[56],[63],[64],[65] These findings are significant because RTKs play a role in the activation of Ras-MAPK signaling and the subsequent phosphorylation of RXRα, which may contribute to liver carcinogenesis. In addition, we have recently reported that ACR inhibits the activation of Ras and the phosphorylation of ERK and RXRα proteins in the liver of DEN-treated obese mice. [49] The inhibitory effects of ACR on Ras activation are also observed in human HCC and pancreatic cancer cells. [52],[66] These findings may indicate that activation of the RTK-Ras-MAPK signaling pathway and subsequent RXRα phosphorylation are critical targets of ACR for the inhibition of HCC development and cancer cell growth [Figure 1].
Chemoprevention of HCC by ACR
In order to test whether ACR can reduce the incidence of recurrent and second primary HCC, an early phase randomized controlled clinical trial was conducted in patients who underwent potentially curative treatment for initial HCC. [8],[9],[10] In this trial, treatment with ACR (administered to 44 patients, 600 mg/day) for 12 months significantly reduced the incidence of recurrent or new HCCs compared with placebo (administered to 45 patients) after a median follow-up period of 38 months; 12 patients (27%) in the ACR group developed HCC compared with 22 patients (49%) in the placebo group (P = 0.04). [8] After further follow-up to 62 months, ACR was also found to improve both recurrence-free survival (P = 0.002) and overall survival (P = 0.04). The estimated 6-year overall survival was 74% in the ACR group and 46% in the placebo group. [9] The relative risks for the development of secondary HCC and death were 0.31 [95% confidence interval (CI), 0.12 – 0.78] and 0.33 (95% CI, 0.11-0.79), respectively. [8],[9] Moreover, the preventive effects of ACR lasted up to 50 months after randomization, or 38 months after completion of the drug. [10] The results of these reports suggest that administration of ACR for only 12 months exerts a long-term effect on the prevention of second primary HCC without causing severe adverse effects from retinoid.
The preventive effect of ACR on the development of second primary HCC in HCV-positive patients, who underwent curative therapy for initial HCC or first recurrence, has also been confirmed by a multicenter large-scale (n = 401) randomized placebo-controlled trial with a median follow-up of 2.5 years. In this trial, oral administration of ACR (600 mg/day) exerted a strong effect on the prevention of second primary HCC with a hazard ratio of 0.27 (95% CI, 0.07-0.96) at 2 years after treatment. Cumulative recurrence-free survival rates in the ACR-treated group were higher than those in the placebo group (after the first year: ACR, 71.9%; placebo, 66.0% and at 3 years: ACR, 43.7%; placebo, 29.3%), indicating that ACR reduced the recurrence of HCC, especially after 2 years of treatment. [67] In addition, subgroup analysis of this study showed the significant result that ACR powerfully prevented the development of a second primary HCC, with a hazard ratio of 0.38 (95% CI, 0.20-0.71) in patients who were Child-Pugh A and had small tumors (size < 20 mm). [68] The results of these clinical trials suggest that ACR inhibits de novo carcinogenesis and is therefore a novel first-line therapy to reduce the development of second primary HCC, especially for patients with well-preserved liver function who have undergone curative therapy for a small tumor. Common treatment-related adverse events observed in this trial were albuminuria, hypertension, and headache; however, these adverse events were tolerated. The safety of ACR was also determined in a phase I pharmacokinetics clinical trial. In that trial, doses of 300 and 600 mg/day did not result in any adverse effects or dose-limiting toxicities, whereas a dose of 900 mg/day resulted in grade 3 hypertension as a dose-limiting toxicity. [69]
Concept of “clonal deletion and inhibition” therapy
The annual incidence of HCC reaches approximately 3% in HBV- and 7% in HCV-infected cirrhotic patients. More serious is that the frequency of HCC recurrence after curative treatment is very high in cirrhotic patients; the annual incidence rises to approximately 20%-25% and the recurrence rate at 5 years after definitive therapy exceeds 70%. [70],[71],[72],[73] This typical clinical course of patients with HCC is associated with the clinical characteristic mode of liver carcinogenesis, multicentric carcinogenesis, which is also expressed by the term “field cancerization.” Once a liver is exposed to a continuous carcinogenic insult, such as hepatitis virus infection, the whole liver is regarded as a precancerous field that possesses multiple as well as independent premalignant or latent malignant clones. Based on this characteristic, a curative treatment for HCC is difficult once this malignancy has developed; therefore, we believe that one of the most promising and practical strategies for HCC treatment is the removal and inhibition of latent malignant clones from the chronically damaged liver that is in a hypercarcinogenic state before the latent malignant clones expand into a clinically detectable tumor. We have proposed this new concept of “clonal deletion and inhibition” therapy for HCC chemoprevention. [74] We believe that ACR prevents the development of HCC through implementation of this concept for the following reasons.
First, the serum levels of lectin-reactive a-fetoprotein factor 3 (AFP-L3) and protein induced by vitamin K absence or antagonist-II (PIVKA-II), both of which indicate the presence of latent (ie, invisible) malignant clones in the remnant liver, were significantly reduced after ACR administration for 12 months in an early-phase clinical trial. [8],[9],[10] Next, ACR was found to prevent the appearance of serum AFP-L3 in patients whose AFP-L3 levels were negative at trial enrolment, whereas the number of patients whose serum AFP-L3 appeared de novo was significantly increased in the placebo group, and these patients had a significantly higher risk of developing secondary HCC. [74] These observations are explained by the implementation of “clonal deletion and inhibition” therapy with ACR; namely, ACR eliminates the AFP-L3- and PIVKA-II-producing premalignant clones from the remnant liver before they expand into clinically detectable tumors (“clonal deletion”). At the same time, ACR inhibits the development of such clones, which have the potential to become HCC, in the liver (“clonal inhibition”). Once the malignant clones are eliminated or inhibited from the remnant liver by ACR, it takes several years for de novo HCC to develop in the cirrhotic liver. Therefore, as demonstrated in an early-phase clinical trial, [10] short-term administration (12 months) of ACR could exert a long-term (ie, several years) preventive effect on HCC development even after termination of treatment. The roles of ACR in the implementation of “clonal deletion and inhibition” therapy are schematically represented in [Figure 2].
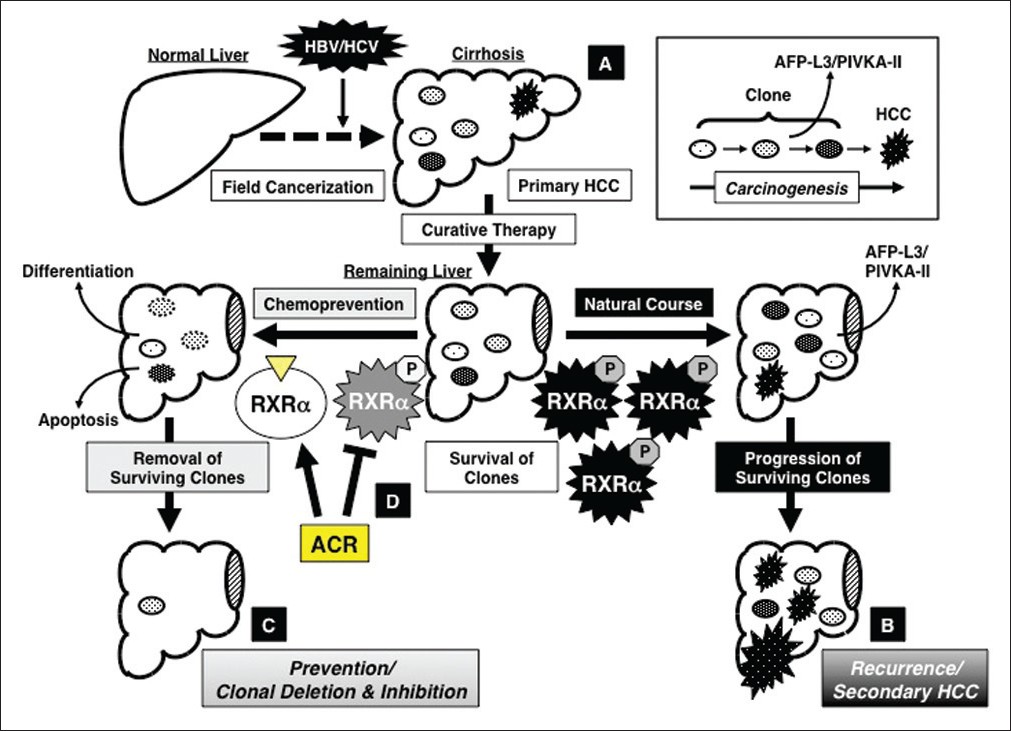 |
Figure 2: Concept of “clonal deletion and inhibition” therapy for hepatocellular carcinoma (HCC) chemoprevention and the effects of acyclic retinoid (ACR) on implementation of this concept. (a) Persistent inflammation caused by hepatitis viral infection transforms the liver into a precancerous field (“field cancerization”), which contains of multiple latent malignant clones that can, at some point, develop into HCC. (b) Even after early detection and removal of the primary HCC, the remaining clones survive in the remaining liver and grow into secondary HCC lesions (natural course), which is a major cause of the poor prognosis for patients with this malignancy. (c) Therefore, one of the most promising strategies to prevent secondary HCC is the deletion and inhibition of such transformed clones by inducing cell differentiation or apoptosis before the clones expand into clinically detectable tumors. This is the concept of “clonal deletion and inhibition” therapy for HCC chemoprevention. (d) ACR, which binds to RXR α and inhibits phosphorylation of this nuclear receptor, prevents the recurrence and development of secondary HCC via the mechanism described by this concept Click here to view |
“Combination chemoprevention” of HCC with ACR
In order to establish more effective strategies to prevent HCC development, we have conducted a study of “combination chemoprevention” using ACR as a key agent. Combination chemoprevention with ACR provides an opportunity to take advantage of the synergistic effects of ACR on growth inhibition in HCC cells. We have initially found that the combination of ACR and interferon-β synergistically inhibits cell growth and induces apoptosis in HCC cells. [58] This finding is significant when considering the clinical use of ACR in the near future because interferon exerts a chemopreventive effect against the recurrence of HCC. [5,6]
In addition to interferon, other agents, particularly those that target RXRα phosphorylation, are also anticipated to be potential partners of ACR for inducing synergistic growth inhibition in HCC cells. For instance, ACR acts synergistically with vitamin K 2 in suppressing growth and inducing apoptosis in human HCC cells by inhibiting Ras-MAPK signaling activation and RXRα phosphorylation. [52] Dephosphorylation of RXRα by targeting the Ras-MAPK signaling pathway and its upstream human epidermal growth factor receptor-2 (HER2) using trastuzumab, a humanized monoclonal antibody against HER2, also enhances the effect of retinoids, including ACR, on inhibiting growth and inducing apoptosis in human HCC cells. [75] Combined treatment with ACR plus valproic acid, a histone deacetylase inhibitor, also acts synergistically to induce apoptosis and G 0 -G 1 cell cycle arrest in HCC cells by inhibiting phosphorylation of RXRα, ERK, Akt, and glycogen synthase kinase-3β proteins. [53] In addition to HCC, in both human pancreatic cancer and leukemia cells, [66],[76] the combination of ACR plus gemcitabine or vitamin K 2 synergistically inhibits cell growth and induces apoptosis by inhibiting Ras activation and RXRα phosphorylation. Moreover, induction of nuclear receptors that dimerize with RXR, such as RAR and PPAR, [77],[78] and recruitment of their ligands also exert synergistic growth inhibition in cancer cells when combined with ACR. [53],[60] In particular, upregulation of cellular levels of RARβ and the subsequent increase of the RARE promoter activity are critical to enhance the ability of ACR to induce apoptosis in the HCC cells. [53],[60]
Conclusion
Finally, we should mention the results of our recent rodent experiment, which showed that ACR has the potential to inhibit obesity-related HCC. [49] In this study, ACR was found to effectively prevent the development of obesity-related liver carcinogenesis by inhibiting the activation of Ras and the phosphorylation of ERK and RXRα in the liver of DEN-treated db/db obese and diabetic mice. [49] This finding is significant because obesity and diabetes mellitus, both of which are major health care problems in the current society, are critical risk factors for HCC development. [2],[79] Therefore, the results of this study may encourage the clinical use of ACR for cirrhotic patients with obesity and diabetes, who are at a notably higher risk for developing HCC.
Retinoids have been used as potential chemotherapeutic or chemopreventive agents because of their differentiation, antiproliferative, and proapoptotic properties. [29],[30] For instance, all-trans-retinoic acid is an effective first-line therapy for the treatment of acute promyelocytic leukemia. [80] We expect that ACR will also become an effective first-line therapy for the prevention of HCC in the near future.
In conclusion, in order to improve the therapeutic outcome for patients with HCC, there is an urgent need to develop more effective strategies for chemoprevention of this malignancy. Realization of the concept of “clonal deletion and inhibition” therapy is one of the most promising and practical approaches for preventing HCC, and the use of ACR is expected to accomplish this goal. Liver carcinogenesis is accompanied by phosphorylation of RXRα, which is a critical target on which ACR can exert its chemopreventive effects against HCC. ACR-based combination chemoprevention, which is based on synergism, also holds great potential for becoming an important strategy for HCC chemoprevention. The clinical application of ACR as an “HCC chemopreventive drug” in patients with liver cirrhosis is awaited with great anticipation.
References
| 1. | Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69-90.  |
| 2. | El-Serag HB, Rudolph KL. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007;132:2557-76. [PUBMED] |
| 3. | Parikh S, Hyman D. Hepatocellular cancer: A guide for the internist. Am J Med 2007;120:194-202. [PUBMED] |
| 4. | Wong JS, Wong GL, Tsoi KK, Wong VW, Cheung SY, Chong CN, et al. Meta-analysis: The efficacy of anti-viral therapy in prevention of recurrence after curative treatment of chronic hepatitis B-related hepatocellular carcinoma. Aliment Pharmacol Ther 2011;33:1104-12. [PUBMED] |
| 5. | Shen YC, Hsu C, Chen LT, Cheng CC, Hu FC, Cheng AL. Adjuvant interferon therapy after curative therapy for hepatocellular carcinoma (HCC): A meta-regression approach. J Hepatol 2010;52:889-94. [PUBMED] |
| 6. | Miyake Y, Takaki A, Iwasaki Y, Yamamoto K. Meta-analysis: Interferon-alpha prevents the recurrence after curative treatment of hepatitis C virus-related hepatocellular carcinoma. J Viral Hepat 2010;17:287-92. [PUBMED] |
| 7. | Muto Y, Sato S, Watanabe A, Moriwaki H, Suzuki K, Kato A, et al. Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long-term oral supplementation with branched-chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol Res 2006;35:204-14. [PUBMED] |
| 8. | Muto Y, Moriwaki H, Ninomiya M, Adachi S, Saito A, Takasaki KT, et al. Prevention of second primary tumors by an acyclic retinoid, polyprenoic acid, in patients with hepatocellular carcinoma. Hepatoma Prevention Study Group. N Engl J Med 1996;334:1561-7. [PUBMED] |
| 9. | Muto Y, Moriwaki H, Saito A. Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. N Engl J Med 1999;340:1046-7. [PUBMED] |
| 10. | Takai K, Okuno M, Yasuda I, Matsushima-Nishiwaki R, Uematsu T, Tsurumi H, et al. Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. Updated analysis of the long-term follow-up data. Intervirology 2005;48:39-45. [PUBMED] |
| 11. | Moriwaki H, Shimizu M, Okuno M, Nishiwaki-Matsushima R. Chemoprevention of liver carcinogenesis with retinoids: Basic and clinical aspects. Hepatol Res 2007;37 Suppl 2:S299-302. [PUBMED] |
| 12. | Shimizu M, Takai K, Moriwaki H. Strategy and mechanism for the prevention of hepatocellular carcinoma: Phosphorylated retinoid X receptor alpha is a critical target for hepatocellular carcinoma chemoprevention. Cancer Sci 2009;100:369-74. [PUBMED] |
| 13. | Shimizu M, Sakai H, Moriwaki H. Chemoprevention of hepatocellular carcinoma by acyclic retinoid. Front Biosci 2011;16:759-69. |
| 14. | Matsushima-Nishiwaki R, Okuno M, Adachi S, Sano T, Akita K, Moriwaki H, et al. Phosphorylation of retinoid X receptor alpha at serine 260 impairs its metabolism and function in human hepatocellular carcinoma. Cancer Res 2001;61:7675-82. [PUBMED] |
| 15. | Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J 1996;10:940-54. |
| 16. | Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, et al. International union of pharmacology. LXIII. Retinoid X receptors. Pharmacol Rev 2006;58:760-72. |
| 17. | Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, et al. International union of pharmacology. LX. Retinoic acid receptors. Pharmacol Rev 2006;58:712-25. |
| 18. | Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International union of pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev 2006;58:726-41. |
| 19. | Okuno M, Kojima S, Akita K, Matsushima-Nishiwaki R, Adachi S, Sano T, et al. Retinoids in liver fibrosis and cancer. Front Biosci 2002;7:d204-18. [PUBMED] |
| 20. | Shirakami Y, Lee SA, Clugston RD, Blaner WS. Hepatic metabolism of retinoids and disease associations. Biochim Biophys Acta 2012;1821:124-36. [PUBMED] |
| 21. | Leo MA, Lieber CS. Hepatic vitamin A depletion in alcoholic liver injury. N Engl J Med 1982;307:597-601. [PUBMED] |
| 22. | Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134:1655-69. [PUBMED] |
| 23. | Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209-18. [PUBMED] |
| 24. | Clemente C, Elba S, Buongiorno G, Berloco P, Guerra V, Di Leo A. Serum retinol and risk of hepatocellular carcinoma in patients with child-Pugh class A cirrhosis. Cancer Lett 2002;178:123-9. [PUBMED] |
| 25. | Newsome PN, Beldon I, Moussa Y, Delahooke TE, Poulopoulos G, Hayes PC, et al. Low serum retinol levels are associated with hepatocellular carcinoma in patients with chronic liver disease. Aliment Pharmacol Ther 2000;14:1295-301. [PUBMED] |
| 26. | Yanagitani A, Yamada S, Yasui S, Shimomura T, Murai R, Murawaki Y, et al. Retinoic acid receptor alpha dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 2004;40:366-75. [PUBMED] |
| 27. | Wan YJ, Cai Y, Lungo W, Fu P, Locker J, French S, et al. Peroxisome proliferator-activated receptor alpha-mediated pathways are altered in hepatocyte-specific retinoid X receptor alpha-deficient mice. J Biol Chem 2000;275:28285-90. [PUBMED] |
| 28. | Gyamfi MA, Tanaka Y, He L, Klaassen CD, Wan YJ. Hepatic effects of a methionine-choline-deficient diet in hepatocyte RXRalpha-null mice. Toxicol Appl Pharmacol 2009;234:166-78. |
| 29. | Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer 2001;1:181-93. [PUBMED] |
| 30. | Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov 2007;6:793-810. [PUBMED] |
| 31. | Muto Y, Moriwaki H. Antitumor activity of vitamin A and its derivatives. J Natl Cancer Inst 1984;73:1389-93. [PUBMED] |
| 32. | Lee JH, Yoon JH, Yu SJ, Chung GE, Jung EU, Kim HY, et al. Retinoic acid and its binding protein modulate apoptotic signals in hypoxic hepatocellular carcinoma cells. Cancer Lett 2010;295:229-35. [PUBMED] |
| 33. | Shirakami Y, Gottesman ME, Blaner WS. Diethylnitrosamine-induced hepatocarcinogenesis is suppressed in lecithin: Retinol acyltransferase-deficient mice primarily through retinoid actions immediately after carcinogen administration. Carcinogenesis 2012;33:268-74. |
| 34. | de The H, Marchio A, Tiollais P, Dejean A. A novel steroid thyroid hormone receptor-related gene inappropriately expressed in human hepatocellular carcinoma. Nature 1987;330:667-70. |
| 35. | Sever CE, Locker J. Expression of retinoic acid alpha and beta receptor genes in liver and hepatocellular carcinoma. Mol Carcinog 1991;4:138-44. [PUBMED] |
| 36. | Li C, Wan YJ. Differentiation and antiproliferation effects of retinoic acid receptor beta in hepatoma cells. Cancer Lett 1998;124:205-11. [PUBMED] |
| 37. | Wan YJ, Wang L, Wu TC. Expression of retinoic acid receptor genes in developing rat livers and hepatoma cells. Lab Invest 1992;66:646-51. [PUBMED] |
| 38. | Ando N, Shimizu M, Okuno M, Matsushima-Nishiwaki R, Tsurumi H, Tanaka T, et al. Expression of retinoid X receptor alpha is decreased in 3¢-methyl-4-dimethylaminoazobenzene-induced hepatocellular carcinoma in rats. Oncol Rep 2007;18:879-84. [PUBMED] |
| 39. | Yan TD, Wu H, Zhang HP, Lu N, Ye P, Yu FH, et al. Oncogenic potential of retinoic acid receptor-gamma in hepatocellular carcinoma. Cancer Res 2010;70:2285-95. |
| 40. | Han YH, Zhou H, Kim JH, Yan TD, Lee KH, Wu H, et al. A unique cytoplasmic localization of retinoic acid receptor-gamma and its regulations. J Biol Chem 2009;284:18503-14. |
| 41. | Garcia AD, Ostapchuk P, Hearing P. Functional interaction of nuclear factors EF-C, HNF-4, and RXR alpha with hepatitis B virus enhancer I. J Virol 1993;67:3940-50. |
| 42. | Matsushima-Nishiwaki R, Shidoji Y, Nishiwaki S, Yamada T, Moriwaki H, Muto Y. Aberrant metabolism of retinoid X receptor proteins in human hepatocellular carcinoma. Mol Cell Endocrinol 1996;121:179-90. |
| 43. | Matsushima-Nishiwaki R, Okuno M, Takano Y, Kojima S, Friedman SL, Moriwaki H. Molecular mechanism for growth suppression of human hepatocellular carcinoma cells by acyclic retinoid. Carcinogenesis 2003;24:1353-9. |
| 44. | Yoshimura K, Muto Y, Shimizu M, Matsushima-Nishiwaki R, Okuno M, Takano Y, et al. Phosphorylated retinoid X receptor alpha loses its heterodimeric activity with retinoic acid receptor beta. Cancer Sci 2007;98:1868-74. |
| 45. | Adachi S, Okuno M, Matsushima-Nishiwaki R, Takano Y, Kojima S, Friedman SL, et al. Phosphorylation of retinoid X receptor suppresses its ubiquitination in human hepatocellular carcinoma. Hepatology 2002;35:332-40. |
| 46. | Moriwaki H, Muto Y, Ninomiya M, Kawai K, Suzuki Y, Seto T. Inhibitory effects of synthetic acidic retinoid and polyprenoic acid on the development of hepatoma in rats induced by 3¢-methyl-N, N-dimethyl-4-aminoazobenzene. Gastroenterol Jpn 1988;23:546-52. |
| 47. | Kagawa M, Sano T, Ishibashi N, Hashimoto M, Okuno M, Moriwaki H, et al. An acyclic retinoid, NIK-333, inhibits N-diethylnitrosamine-induced rat hepatocarcinogenesis through suppression of TGF-alpha expression and cell proliferation. Carcinogenesis 2004;25:979-85. |
| 48. | Sano T, Kagawa M, Okuno M, Ishibashi N, Hashimoto M, Yamamoto M, et al. Prevention of rat hepatocarcinogenesis by acyclic retinoid is accompanied by reduction in emergence of both TGF-alpha-expressing oval-like cells and activated hepatic stellate cells. Nutr Cancer 2005;51:197-206. |
| 49. | Shimizu M, Sakai H, Shirakami Y, Iwasa J, Yasuda Y, Kubota M, et al. Acyclic retinoid inhibits diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BLKS/J- +(db)/+Lepr(db) mice. Cancer Prev Res (Phila) 2011;4:128-36. |
| 50. | Araki H, Shidoji Y, Yamada Y, Moriwaki H, Muto Y. Retinoid agonist activities of synthetic geranyl geranoic acid derivatives. Biochem Biophys Res Commun 1995;209:66-72. |
| 51. | Yamada Y, Shidoji Y, Fukutomi Y, Ishikawa T, Kaneko T, Nakagama H, et al. Positive and negative regulations of albumin gene expression by retinoids in human hepatoma cell lines. Mol Carcinog 1994;10:151-8. |
| 52. | Kanamori T, Shimizu M, Okuno M, Matsushima-Nishiwaki R, Tsurumi H, Kojima S, et al. Synergistic growth inhibition by acyclic retinoid and vitamin K2 in human hepatocellular carcinoma cells. Cancer Sci 2007;98:431-7. |
| 53. | Tatebe H, Shimizu M, Shirakami Y, Sakai H, Yasuda Y, Tsurumi H, et al. Acyclic retinoid synergises with valproic acid to inhibit growth in human hepatocellular carcinoma cells. Cancer Lett 2009;285:210-7. |
| 54. | Fukutomi Y, Omori M, Muto Y, Ninomiya M, Okuno M, Moriwaki H. Inhibitory effects of acyclic retinoid (polyprenoic acid) and its hydroxy derivative on cell growth and on secretion of alpha-fetoprotein in human hepatoma-derived cell line (PLC/PRF/5). Jpn J Cancer Res 1990;81:1281-5. |
| 55. | Nakamura N, Shidoji Y, Yamada Y, Hatakeyama H, Moriwaki H, Muto Y. Induction of apoptosis by acyclic retinoid in the human hepatoma-derived cell line, HuH-7. Biochem Biophys Res Commun 1995;207:382-8. |
| 56. | Nakamura N, Shidoji Y, Moriwaki H, Muto Y. Apoptosis in human hepatoma cell line induced by 4,5-didehydro geranylgeranoic acid (acyclic retinoid) via down-regulation of transforming growth factor-alpha. Biochem Biophys Res Commun 1996;219:100-4. |
| 57. | Yasuda I, Shiratori Y, Adachi S, Obora A, Takemura M, Okuno M, et al. Acyclic retinoid induces partial differentiation, down-regulates telomerase reverse transcriptase mRNA expression and telomerase activity, and induces apoptosis in human hepatoma-derived cell lines. J Hepatol 2002;36:660-71. |
| 58. | Obora A, Shiratori Y, Okuno M, Adachi S, Takano Y, Matsushima-Nishiwaki R, et al. Synergistic induction of apoptosis by acyclic retinoid and interferon-beta in human hepatocellular carcinoma cells. Hepatology 2002;36:1115-24. |
| 59. | Suzui M, Masuda M, Lim JT, Albanese C, Pestell RG, Weinstein IB. Growth inhibition of human hepatoma cells by acyclic retinoid is associated with induction of p21(CIP1) and inhibition of expression of cyclin D1. Cancer Res 2002;62:3997-4006. |
| 60. | Shimizu M, Suzui M, Deguchi A, Lim JT, Xiao D, Hayes JH, et al. Synergistic effects of acyclic retinoid and OSI-461 on growth inhibition and gene expression in human hepatoma cells. Clin Cancer Res 2004;10:6710-21. |
| 61. | Suzui M, Shimizu M, Masuda M, Lim JT, Yoshimi N, Weinstein IB. Acyclic retinoid activates retinoic acid receptor beta and induces transcriptional activation of p21(CIP1) in HepG2 human hepatoma cells. Mol Cancer Ther 2004;3:309-16. |
| 62. | Tatsukawa H, Sano T, Fukaya Y, Ishibashi N, Watanabe M, Okuno M, et al. Dual induction of caspase 3- and transglutaminase-dependent apoptosis by acyclic retinoid in hepatocellular carcinoma cells. Mol Cancer 2011;10:4. |
| 63. | Komi Y, Sogabe Y, Ishibashi N, Sato Y, Moriwaki H, Shimokado K, et al. Acyclic retinoid inhibits angiogenesis by suppressing the MAPK pathway. Lab Invest 2010;90:52-60. |
| 64. | Shimizu M, Suzui M, Deguchi A, Lim JT, Weinstein IB. Effects of acyclic retinoid on growth, cell cycle control, epidermal growth factor receptor signaling, and gene expression in human squamous cell carcinoma cells. Clin Cancer Res 2004;10:1130-40. |
| 65. | Shao RX, Otsuka M, Kato N, Taniguchi H, Hoshida Y, Moriyama M, et al. Acyclic retinoid inhibits human hepatoma cell growth by suppressing fibroblast growth factor-mediated signaling pathways. Gastroenterology 2005;128:86-95. |
| 66. | Nakagawa T, Shimizu M, Shirakami Y, Tatebe H, Yasuda I, Tsurumi H, et al. Synergistic effects of acyclic retinoid and gemcitabine on growth inhibition in pancreatic cancer cells. Cancer Lett 2009;273:250-6. |
| 67. | Okita K, Matsui O, Kumada H, Tanaka K, Kaneko S, Moriwaki H, et al. Effect of peretinoin on recurrence of hepatocellular carcinoma (HCC): Results of a phase II/III randomized placebo-controlled trial. J Clin Oncol 2010;28 Suppl 15:abstr 4024. |
| 68. | Okusaka T, Makuuchi M, Matsui O, Kumada H, Tanaka K, Kaneko S, et al. Clinical benefit of peretinoin for the suppression of hepatocellular carcinoma (HCC) recurrence in patients with Child-Pugh grade A (CP-A) and small tumor: A subgroup analysis in a phase II/III randomized, placebo-controlled trial. J Clin Oncol 2011, 29 Suppl 4: abstr 165. |
| 69. | Okusaka T, Ueno H, Ikeda M, Morizane C. Phase I and pharmacokinetic clinical trial of oral administration of the acyclic retinoid NIK-333. Hepatol Res 2011;41:542-52. |
| 70. | Kumada T, Nakano S, Takeda I, Sugiyama K, Osada T, Kiriyama S, et al. Patterns of recurrence after initial treatment in patients with small hepatocellular carcinoma. Hepatology 1997;25:87-92. |
| 71. | Koda M, Murawaki Y, Mitsuda A, Ohyama K, Horie Y, Suou T, et al. Predictive factors for intrahepatic recurrence after percutaneous ethanol injection therapy for small hepatocellular carcinoma. Cancer 2000;88:529-37. |
| 72. | Tsukuma H, Hiyama T, Tanaka S, Nakao M, Yabuuchi T, Kitamura T, et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med 1993;328:1797-801. |
| 73. | Okada S, Shimada K, Yamamoto J, Takayama T, Kosuge T, Yamasaki S, et al. Predictive factors for postoperative recurrence of hepatocellular carcinoma. Gastroenterology 1994;106:1618-24. |
| 74. | Moriwaki H, Yasuda I, Shiratori Y, Uematsu T, Okuno M, Muto Y. Deletion of serum lectin-reactive alpha-fetoprotein by acyclic retinoid: A potent biomarker in the chemoprevention of second primary hepatoma. Clin Cancer Res 1997;3:727-31. |
| 75. | Tatebe H, Shimizu M, Shirakami Y, Tsurumi H, Moriwaki H. Synergistic growth inhibition by 9-cis-retinoic acid plus trastuzumab in human hepatocellular carcinoma cells. Clin Cancer Res 2008;14:2806-12. |
| 76. | Kitagawa J, Hara T, Tsurumi H, Ninomiya S, Ogawa K, Adachi S, et al. Synergistic growth inhibition in HL-60 cells by the combination of acyclic retinoid and vitamin K2. J Cancer Res Clin Oncol 2011;137:779-87. |
| 77. | Yamazaki K, Shimizu M, Okuno M, Matsushima-Nishiwaki R, Kanemura N, Araki H, et al. Synergistic effects of RXR alpha and PPAR gamma ligands to inhibit growth in human colon cancer cells-phosphorylated RXR alpha is a critical target for colon cancer management. Gut 2007;56:1557-63. |
| 78. | Shimizu M, Moriwaki H. Synergistic effects of PPARgamma ligands and retinoids in cancer treatment. PPAR Res 2008;2008:181047. |
| 79. | El-Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004;126:460-8. |
| 80. | Kamimura T, Miyamoto T, Harada M, Akashi K. Advances in therapies for acute promyelocytic leukemia. Cancer Sci 2011;102:1929-37. |
Authors
Dr. Koji Takai, Department of Gastroenterology/Internal Medicine, Gifu University Graduate School of Medicine 1-1 Yanagido, Gifu, Japan 501-1194.
Dr. Hisataka Moriwaki, Department of Gastroenterology/Internal Medicine, Gifu University Graduate School of Medicine 1-1 Yanagido, Gifu, Japan 501-1194.
Dr. Masahito Shimizu, Department of Gastroenterology/Internal Medicine, Gifu University Graduate School of Medicine 1-1 Yanagido, Gifu, Japan 501-1194.
Dr. Yohei Shirakami, Department of Gastroenterology/Internal Medicine, Gifu University Graduate School of Medicine 1-1 Yanagido, Gifu, Japan 501-1194.
Dr. Kenji Imai, Department of Gastroenterology/Internal Medicine, Gifu University Graduate School of Medicine 1-1 Yanagido, Gifu, Japan 501-1194.
Figures