Takeo Shimasaki1, Ayako Kitano2, Yoshiharu Motoo3, Toshinari Minamoto2
1 Department of Advanced Medicine, Medical Research Institute, Kanazawa Medical University and Hospital, 1-1 Daigaku, Uchinada, Kahoku-gun, Ishikawa 920-0293; Division of Translational and Clinical Oncology, Cancer Research Institute and Cancer Center, Kanazawa University and Hospital, 13-1 Takara-machi, Kanazawa 920-0934, Japan
2 Division of Translational and Clinical Oncology, Cancer Research Institute and Cancer Center, Kanazawa University and Hospital, 13-1 Takara-machi, Kanazawa 920-0934, Japan
3 Department of Medical Oncology, Kanazawa Medical University and Hospital, 1-1 Daigaku, Uchinada, Kahoku-gun, Ishikawa 920-0293, Japan
| Date of Submission | 20-Mar-2012 |
| Date of Acceptance | 15-Jul-2012 |
| Date of Web Publication | 13-Sep-2012 |
Correspondence Address:
Takeo Shimasaki
Department of Advanced Medicine, Medical Research Institute, Kanazawa Medical University and Hospital, 1-1 Daigaku, Uchinada, Kahoku-gun, Ishikawa 920-0293; Division of Translational and Clinical Oncology, Cancer Research Institute and Cancer Center, Kanazawa University and Hospital, 13-1 Takara-machi, Kanazawa 920-0934
Japan
Source of Support: None, Conflict of Interest: None
DOI: 10.4103/1477-3163.100866
Abstract
Development and progression of pancreatic cancer involves general metabolic disorder, local chronic inflammation, and multistep activation of distinct oncogenic molecular pathways. These pathologic processes result in a highly invasive and metastatic tumor phenotype that is a major obstacle to curative surgical intervention, infusional gemcitabine-based chemotherapy, and radiation therapy. Many clinical trials with chemical compounds and therapeutic antibodies targeting growth factors, angiogenic factors, and matrix metalloproteinases have failed to demonstrate definitive therapeutic benefits to refractory pancreatic cancer patients. Glycogen synthase kinase 3β (GSK3β), a serine/threonine protein kinase, has emerged as a therapeutic target in common chronic and progressive diseases, including cancer. Here we review accumulating evidence for a pathologic role of GSK3β in promoting tumor cell survival, proliferation, invasion, and resistance to chemotherapy and radiation in pancreatic cancer. We also discuss the putative involvement of GSK3β in mediating metabolic disorder, local inflammation, and molecular alteration leading to pancreatic cancer development. Taken together, we highlight potential therapeutic as well as preventive effects of GSK3β inhibition in pancreatic cancer.
Keywords: Carcinogenesis, GSK3β, pancreatic cancer, tumor progression
How to cite this article:
Shimasaki T, Kitano A, Motoo Y, Minamoto T. Aberrant glycogen synthase kinase 3β in the development of pancreatic cancer. J Carcinog 2012;11:15
How to cite this URL:
Shimasaki T, Kitano A, Motoo Y, Minamoto T. Aberrant glycogen synthase kinase 3β in the development of pancreatic cancer. J Carcinog [serial online] 2012 [cited 2021 Oct 14];11:15. Available from: https://carcinogenesis.com/text.asp?2012/11/1/15/100866
Introduction
Pancreatic cancer (referred hereafter to pancreatic ductal adenocarcinoma as the major cancer subtype in the pancreas) is one of the most common malignant and lethal tumors, and a major unsolved health problem due to its late clinical diagnosis and predisposition toward metastasis. [1],[2],[3] Most patients with advanced-stage pancreatic cancer have little opportunity to undergo curative surgery. [4] Chemotherapy and radiotherapy, either alone or in combination, are the primary palliative treatment options available for these patients. Despite substantial treatment advances, [5] the prognosis of pancreatic cancer patients is still unfavorable. [6],[7] Understanding the detailed molecular and biologic basis of pancreatic cancer pathogenesis facilitates advances in treatment and prevention.
Pancreatic cancer develops through cumulative biologic processes involving metabolic disorder [8],[9] and chronic local inflammation [10],[11] in association with host stromal changes [12],[13] and deregulated molecular pathways. [14],[15],[16],[17] New therapeutic strategies that target growth factors, angiogenic factors, and matrix metalloproteinases have been developed based on molecular mechanisms. [14],[18] Small-molecule agents and therapeutic monoclonal antibodies targeting these factors are currently available, but many clinical trials have failed to demonstrate definitive therapeutic benefits of these molecular-targeted approaches for refractory pancreatic cancer patients. [19] Therefore, identification of novel molecular targets that can enhance the effects of conventional chemotherapy and radiation is a high priority. [20]
Glycogen synthase kinase (GSK)3β is a serine/threonine protein kinase that has emerged as a therapeutic target for chronic and progressive diseases, including cancer. [21],[22],[23],[24] Here we review studies that investigated the pathologic role of GSK3β in tumor cell survival, proliferation, invasion, and resistance to chemotherapy and radiation in pancreatic cancer. We discuss the putative involvement of GSK3β in mediating systemic metabolic disorder, local inflammation, and molecular alteration leading to pancreatic cancer development, and highlight the potential therapeutic as well as preventive effects of GSK3β inhibition in pancreatic cancer.
Biology and Pathology of Gsk3β
GSK3 was discovered in 1980 as a protein kinase that phosphorylates glycogen synthase (GS), a key enzyme involved in glycogen synthesis under the control of insulin signalling. [25] GSK3 is evolutionarily conserved and consists of two distinct isoforms encoded by different genes in mammals, GSK3α and GSK3β. [26] GSK3α has an additional 60 amino acids at the N-terminus, but the kinase domains of the two isoforms are highly homologous. Unlike most protein kinases, GSK3b is active in normal cells. [2] Its activity is controlled by subcellular localization, different binding partners, and differential phosphorylation at serine 9 (S9; inactive form) and tyrosine 216 (Y216; active form).
GSK3β is primarily a negative regulator of glycogen synthesis through phosphorylation and inactivation of GS, but it also participates in cell cycle regulation, proliferation, differentiation, apoptosis, and migration. [27],[28],[29] Based on its causative associations with glucose intolerance, neuronal degeneration, and inflammation, GSK3β is a therapeutic target for common chronic diseases, such as type 2 diabetes mellitus (DM) and Alzheimer’s disease. [21],[22],[23] The osteogenic function of Wnt/β-catenin signaling [30],[31],[32] suggests that GSK3β could be a therapeutic target for osteoporotic bone disease. An orally bioavailable GSK3α/β dual inhibitor was generated and tested as a new drug for osteoporosis treatment [33] based on the role of GSK3β as part of a complex involved in ubiquitin-mediated proteasomal degradation of β-catenin. [34],[35] GSK3β also upregulates basal gastric acid secretion, [36] suggesting a causative role in peptic ulcer diseases. Thus, GSK3β is being increasingly recognized as an attractive drug target for an expanding spectrum of chronic diseases. [37]
Under physiological conditions, GSK3β targets several transcription factors, cell cycle regulators, and proto-oncoproteins for phosphorylation and subsequent degradation via the ubiquitin-proteasome system. Therefore, it is hypothesized that GSK3β suppresses tumor development by attenuating certain oncogenic signaling cascades mediated by Wnt and hedgehog. [38],[39] There is, however, little experimental evidence showing GSK3β inactivation or loss of its expression during tumor development and progression.
An increase in GSK3β expression, its active form (phosphorylated at Y216) and activity, and a decrease in its inactive form (phosphorylated at S9) are distinct features of gastrointestinal cancers, including pancreatic cancer, and glioblastoma. GSK3β sustains survival and proliferation of these tumor cells, and pharmacologic inhibition of its activity reduces their survival and proliferation, and predisposes them to apoptosis in vitro and in tumor xenografts. [40],[41],[42],[43],[44] Consequences of GSK3β inhibition in cancer cells include restoration of p53- and Rb-mediated pathways, and downregulation of human telomerase reverse transcriptase and telomerase leading to cell senescence. [42],[43] With these observations, we have proposed GSK3β as a potential target for cancer treatment. [24],[45] Although its role in cancer is still debated, [46] the overall results thus far indicate that aberrant expression and activity of GSK3β is a common and fundamental characteristic in a broad spectrum of cancers. [24]
Putative Involvement of Gsk3β in Pancreatic Cancer Development
There is accumulating epidemiologic evidence that persistent glucose intolerance and chronic local inflammation are risk factors for pancreatic cancer. [8],[47] Characteristic molecular alterations during pancreatic cancer development and progression include frequent mutational activation of the K-ras oncoprotein and infrequent activation of Wnt/β-catenin signaling. [14],[15],[16],[48],[49] Here we discuss the putative participation of GSK3β in mediating these epidemiologic risk factors and oncogenic signaling pathways leading to pancreatic cancer development [Figure 1].
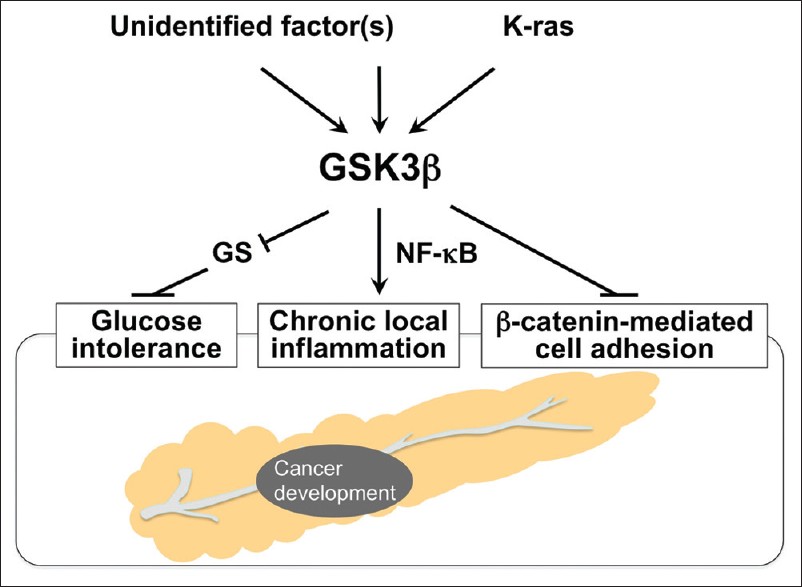 |
Figure 1: Systemic and local effects of deregulated GSK3 b on known risk factors for pancreatic carcinogenesis Click here to view |
Glucose intolerance and pancreatic cancer
The association between glucose intolerance and pancreatic cancer risk has long been a major epidemiologic issue of interest. Recent reviews and meta-analyses of prospective observational studies have demonstrated that obesity is significantly associated with an increased risk of pancreatic cancer development. [50],[51] DM is also a clinical manifestation of pancreatic cancer, and well-designed case-control and prospective studies have demonstrated an increased risk of pancreatic cancer in patients with long-term DM, [52],[53],[54] suggesting the causative association between the two diseases. Consistently, a number of studies have reported the potential role of glucose-lowering therapies in reducing the risk of pancreatic cancer. [55] Metformin, for example, is a biguanide compound that is prescribed for approximately a hundred million patients with DM. [56] A comprehensive review and meta-analysis of epidemiologic studies demonstrated an inverse correlation between the use of this drug and incidence of pancreatic cancer. [57] The primary function of GSK3β is to phosphorylate and inactivate GS, and thus attenuate conversion of glucose to glycogen. This implies that deregulated GSK3β will indirectly facilitate pancreatic carcinogenesis by affecting systemic glucose metabolism [Figure 1].
Chronic pancreatitis and pancreatic cancer
The observed association of chronic pancreatitis with increased risk of developing pancreatic cancer [47],[58],[59] is supported by a recent meta-analysis of 22 well-performed epidemiologic studies. [10] The incidence of chronic pancreatitis in the general population estimated from hospitalization data is only 5-10 per 100,000 persons a year. Although the risk of developing pancreatic cancer is much higher for patients with hereditary pancreatitis than for patients with sporadic chronic pancreatitis, [60],[61],[62] the incidence of hereditary pancreatitis due to a germline mutation in the cationic trypsinogen gene [63] is less than 1% of all chronic pancreatitis cases. Accordingly, chronic pancreatitis is not a precursor to pancreatic cancer in a majority of cases.
Despite such a low incidence, both forms of chronic pancreatitis have presented substantial evidence for putative inflammatory mechanisms contributing to pancreatic cancer development and progression. These mechanisms involve pro-inflammatory cytokines, nuclear factor (NF)-κB, cyclooxygenase-2, peroxisome proliferator-activated receptor-γ, nitric oxide (NO) synthesized by inducible NO synthase, DNA damage caused by release of proteolytic enzymes and reactive oxygen species as well as somatic mutations in oncogenes (eg, K-ras), and tumor suppressor genes (eg, p53, p16, DPC/Smad). [64],[65],[66] Notably, previous studies of gene knockout mice showed that the expression and activity of GSK3β are crucial for normal cell survival mediated by the NF-κB pathway. [67],[68] Given its pro-inflammatory role, [23] GSK3β may contribute to pancreatic carcinogenesis by promoting chronic local inflammation in the pancreas [Figure 1]. Pancreatic cancer progression shares these molecular alterations, which are promising targets for early molecular diagnosis, treatment, and prevention of the disease. [11],[64],[65],[66]
K-ras and GSK3β cooperate in pancreatic cancer development
K-ras and β-catenin mediate the major oncogenic signaling pathways during gastrointestinal carcinogenesis. [34],[35],[49],[69] GSK3β is overexpressed and constitutively active in a number of cancer types, including colorectal and pancreatic
cancers. [40],[42],[43],[44],[70],[71],[72],[73] A previous study of colon cancer cells reported that the K-ras oncoprotein transactivates β-catenin via inactivation of GSK3β. [74] However, this is inconsistent with our studies showing that K-ras and β-catenin are independently activated, but do not interact. [75] There is no evidence of a straightforward association between β-catenin activation and levels of GSK3β expression or activity in clinical colorectal cancers. [40],[43] The latter observation is supported by evidence that β-catenin is activated in most cases of colorectal cancer by truncation of the tumor suppressor, adenomatous polyposis coli (APC), or by mutation of the phospho-acceptor sites (S33, S37, and T41 residues) of the CTNNB1 gene encoding β-catenin in cases with wild-type APC. [34],[49]
Mutational activation of the K-ras proto-oncogene is the most prominent event [77] among the deregulated oncogenic pathways leading to pancreatic carcinogenesis. [15],[16] Interestingly, a recent study has indicated that mutant K-ras-mediated signaling increases GSK3β expression in pancreatic cancer cells via promotion of its transcription by the canonical mitogen-activated protein kinase signaling pathway. [78] This finding may suggest that the activated K-ras and GSK3β pathways cooperate to facilitate development and progression of pancreatic cancer [Figure 1].
β-catenin as a bystander in pancreatic cancer: A consequence of deregulated GSK3β?
In contrast to the frequent mutational activation of K-ras, neither mutations in the CTNNB1 phospho-acceptor sites (S33, S37, T41 residues) nor nuclear translocation of (stabilized) β-catenin have been detected in human pancreatic cancer, [48],[79] except in rare cases of pancreatic solid-pseudopapillary tumor. [80] These data suggest that β-catenin is a “bystander” that plays no critical role in tumorigenesis or progression of pancreatic cancer [34] although this remains controversial. [49] It is intriguing to hypothesize that if GSK3β retains its function as part of the canonical degradation machinery of β-catenin [34],[35] in tumor cells, a lack of β-catenin activation may be a consequence of GSK3β overexpression and increased activity in pancreatic cancers. Otherwise, deregulated GSK3β may dissociate E-cadherin-mediated cell adhesion complexes [81] via destabilization of β-catenin, thereby promoting pancreatic carcinogenesis [Figure 1].
Pivotal Roles of Aberrant Gsk3β in Pancreatic Cancer Progression
As in all cancers, unrestrained cell survival and proliferation predominantly contribute to the progression of pancreatic cancer. In this section, we address the pivotal roles of deregulated GSK3β in pancreatic cancer and the underlying biologic mechanisms.
Cancer cell survival and proliferation
The distinct pathologic role of GSK3β in sustaining and promoting tumor cell survival and proliferation is supported by observed effects of GSK3β inhibition against pancreatic cancer cells and their xenografts. [43],[44],[70],[82],[83] Based on studies showing the crucial role for GSK3β in normal cell survival mediated by the NF-κB pathway, [67],[68] GSK3β was also found to support survival of pancreatic cancer cells via NF-κB transcriptional activity. [82],[83] The effect of GSK3β inhibition against pancreatic cancer cells is associated with induction of p53- and Rb-mediated pathways. [43] Taken together, GSK3β sustains tumor cell survival via modulation of both oncogenic and tumor suppressor pathways involved in pancreatic tumorigenesis [Figure 2]. GSK3β also contributes to the progression of various cancer types by modulating cyclin D1, cyclin-dependent kinases (CDKs), c-Myc and p27 Kip1 . [24],[45]
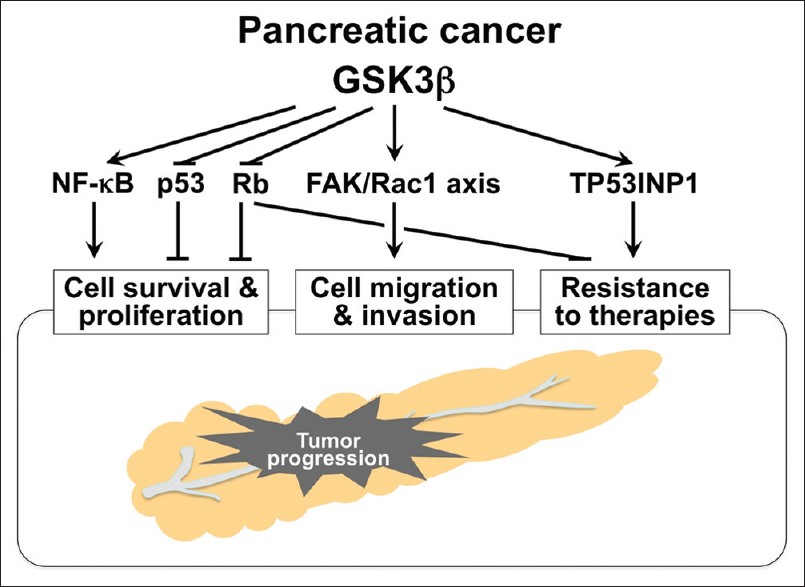 |
Figure 2: Multidirectional effects of deregulated GSK3 b on pancreatic cancer progression and underlying molecular mechanisms Click here to view |
Cancer cell invasion
The highly invasive phenotype characteristic of pancreatic cancer cells not only represents a biological property of tumor progression, but also a major obstacle to curative surgical intervention, chemotherapy and radiation. The proinvasive phenotype of cancer cells is associated with distinct morphological and functional changes that mimic mesenchymal cells, designated as the epithelial-mesenchymal transition (EMT), and increased cell motility. [84],[85] GSK3β inhibits EMT by phosphorylating and destabilizing snail, a transcriptional repressor of E-cadherin. [86] However, in our preliminary study, GSK3β inhibition did not induce the morphological or functional changes responsible for EMT in pancreatic cancer cells (unpublished observation). A possible explanation may be that the established pancreatic cancer cell lines have already acquired the EMT phenotype.
GSK3β is a key regulator of cell polarization and migration during physiological processes such as tissue development and wound healing. [87] Very little is known about its role in the migration and invasion of cancer cells. We recently found that GSK3β inhibition attenuates chemotactic migration and invasion of pancreatic cancer cells. This effect is associated with alterations in subcellular localization of Rac-1 and F-actin, and in cellular microarchitecture, including lamellipodia (unpublished observation), a characteristic morphological change responsible for cell migration. [85] EMT may not be involved in the mechanism by which GSK3β inhibition attenuates pancreatic cancer cell migration and invasion (unpublished observation). Rac-1 destabilizes E-cadherin-mediated cell adhesion in pancreatic cancer cells. [88] These findings indicate that deregulated GSK3β enhances pancreatic cancer cell invasion by inhibiting cell adhesion, and by inducing the morphological and functional changes responsible for cell migration and invasion [Figure 2]. Further work is required to clarify the putative roles for GSK3β in regulating cytoskeletal structure, cell polarity and motility, and hence its promotion of cancer cell migration and invasion.
Potential Influence of Gsk3β on Cancer Cell Susceptibility to Therapy
The intrinsic and acquired resistance of pancreatic cancer to conventional chemotherapy and radiation therapy further facilitates tumor progression, and thus represents a major obstacle to effective treatment of this disease. Novel therapeutic strategies are urgently needed to enhance the effects of these conventional therapies, as well as to attenuate the highly invasive properties of pancreatic cancer cells as discussed above.
Current status of therapeutic options for pancreatic cancer
As mentioned, the aggressive biological nature of pancreatic cancer is an obstacle to early diagnosis and curative surgical intervention. Infusional gemcitabine is the current chemotherapy for patients with unresectable or recurrent pancreatic cancer, although fewer than 20% of patients respond to this treatment. [1],[2],[3],[89] Radiation therapy alone or in combination with gemcitabine-based chemotherapy has a role in the treatment of locally advanced pancreatic cancer (eg, stage III), whereas its role in stage IV (metastatic) disease is palliative. [1],[2],[90],[91] Radiation therapy as pre- or post-operative treatment invokes controversy, and these are areas under study. Two groups of investigators proposed a new combination chemotherapeutic regimen, FOLFIRINOX (leucovorin, 5-fluorouracil, irinotecan, and oxaliplatin), and tested its effects on metastatic pancreatic cancer compared to gemcitabine. They found that it was associated with increased survival and toxicity, concluding that FOLFIRINOX is a treatment option for metastatic pancreatic cancer patients with good performance status, [92] whereas it was considered as an advance in the light of numerous negative phase III clinical trials described below. The need for development of strategies for enhancing anti-tumor effects of gemcitabine and radiation is urgent and important to reverse refractory pancreatic cancer.
Molecular target-directed therapy has emerged as a biology-based treatment modality. For pancreatic cancer treatment, this approach includes targeting of the epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF) and platelet-derived growth factor receptor (PDGFR), since these are aberrantly expressed in pancreatic cancer. [14],[18] Currently available agents targeting these molecules include anti-EGFR antibodies (cetuximab, panitumumab), small-molecule EGFR inhibitors (gefitinib, erlotinib), an anti-VEGF antibody (bevacizumab) and a small-molecule VEGF receptor (VEGFR) inhibitor (axitinib). A number of phase III clinical trials for treating pancreatic cancer patients have been conducted using a single molecular-targeted agent alone or in combination with gemcitabine. These trials have shown little therapeutic benefit to the patients enrolled [19] , with the exception of a combination of erlotinib and gemcitabine, which shows only a marginally significant benefit. [93] Therefore, identification of new molecular target(s) as well as approach for modulation of tumor microenvironment (eg, tumor stroma, [12],[13],[94] tumor-associated inflammation [65],[66] ) is necessary for developing therapeutic strategies that enhance the effect of gemcitabine and improve patient survival.
Implication of GSK3β in pancreatic cancer resistance to therapies
There have been many attempts to investigate the molecular and biological mechanisms by which pancreatic cancer cells resist or acquire resistance to chemotherapy and radiation, focusing mainly on NF-κB. [95] An effective way to overcome pancreatic cancer cell resistance to these therapies has not been identified clinically. GSK3β sustains pancreatic cancer cell survival by maintaining the transcriptional activity of NF-κB. [82],[83] However, a previous study showed that disruption of NF-κB transcription activity through GSK3β inhibition did not sensitize pancreatic cancer cells to gemcitabine. [96] Whereas these studies examined activity of exogenous (transfected) NF-κB, we found no effect of GSK3β inhibition on endogenous NF-κB transcriptional activity in gastrointestinal cancers, including pancreatic cancer. [42],[43] Therefore, a role for GSK3β in modulating NF-κB activity in pancreatic cancer cells remains controversial.
We recently tested various combinations and doses of gemcitabine and a small-molecule GSK3β inhibitor (AR-A014418) on pancreatic cancer cell survival, and demonstrated that the GSK3β inhibitor significantly and synergistically enhances the effect of gemcitabine against cancer cells and their xenograft tumors when the doses and treatment protocol were optimized. [44] cDNA microarray-based comparison of gene expression profiles between sham-treated cancer cells and cells treated with gemcitabine alone or in combination with AR-A014418 showed that the GSK3β inhibitor substantially decreases the level of tumor protein 53-induced nuclear protein 1 (TP53INP1) expression upregulated by gemcitabine treatment. [44] TP53INP1 is identical to thymus-expressed acidic protein, stress-induced protein and p53-dependent damage-inducible nuclear protein 1, functions to repair stress-induced DNA damage [97],[98],[99] and is a critical tumor suppressor in pancreatic cancer. [100] The biological properties of TP53INP1 suggest that the distinct DNA damage repair machinery confers pancreatic cancer cell resistance to gemcitabine [Figure 2].
Another possible mechanism by which GSK3β renders pancreatic cancer cells chemoresistant is suggested by our preliminary observation that GSK3β inhibition decreases Rb phosphorylation (unpublished), thus restoring its function against E2F. E2F transcriptional targets include ribonucleotide reductase (RR), thymidylate synthase and thymidine kinase. These enzymes are essential for DNA synthesis and replication, [101] and pancreatic cancers with increased RR expression are resistant to gemcitabine. [102] These data suggest that activation of Rb following GSK3β inhibition and the resultant inactivation of E2F may sensitize cancer cells to gemcitabine by affecting RR expression [Figure 2]. Consequently, GSK3β inhibition combined with chemotherapy is a novel and promising strategy for sensitizing pancreatic cancer cells to gemcitabine.
EMT induction in cancer cells is associated with acquisition of resistance to chemotherapeutic agents in several cancer types, including pancreatic cancer. [103],[104],[105],[106] The underlying mechanism involves regulators of EMT thought to associate with and maintain acquired chemoresistance in pancreatic cancer cells. Given the role of GSK3β in cancer cell invasion and resistance to therapies as discussed above, it is of particular interest to address whether deregulated GSK3β participates in acquisition of EMT-mediated chemoresistance in cancer cells.
Perspectives
We provided an overview of the role of GSK3β in development and progression of pancreatic cancer, and discussed the underlying molecular and biological mechanisms based on findings from available studies and our preliminary observations. In particular, the indirect contribution of GSK3β to pancreatic carcinogenesis through its causative influence on general metabolic disorder and local chronic inflammation [Figure 1] highlights the importance of exploring the potential chemopreventive effects of GSK3β inhibition against pancreatic cancer as well as other cancer types that depend on GSK3β for survival and proliferation.
There are concerns regarding the therapeutic and chemopreventive use of GSK3β inhibitors because they may activate oncogenic (eg, Wnt, hedgehog) signaling and thus promote cellular transformation. [38],[39],[46] It is well documented, however, that GSK3β inhibition is not sufficient to stabilize β-catenin in normal cells and this appears to occur only when one or more transforming events, such as APC protein truncation, has already taken place. [107] Furthermore, the critical function of GSK3β in mediating Wnt/β-catenin signaling in normal cells is performed by cell membrane-associated GSK3β. This antagonizes the phosphorylation of β-catenin by cytoplasmic GSK3β and thus its degradation. [108] GSK3β also positively regulates hedgehog signaling through Sufu in mammalian cells. [109] These findings contradict the hypothetical tumor suppressor function of GSK3β. [39]
Several studies have suggested opposite roles for GSK3β in the same cellular events between non-neoplastic and cancer cells. [24] One example involves the CDK inhibitor p27 Kip1 that normally regulates the cell cycle by binding and inactivating the cyclin A/E-CDK2 complex. Whereas GSK3β phosphorylates and stabilizes p27 Kip1 in normal cells, [110] it down-regulates p27 Kip1 in leukemia cells and thus selectively maintains survival and proliferation of these cells. [111] Other studies have reported various roles for GSK3β in regulating cell stemness. GSK3β inhibition maintains embryonic stem cell pluripotency and the repopulates hematopoietic stem cells through activation of the Wnt and hedgehog pathways, respectively. [112],[113] Conversely, GSK3β sustains tumor cell stemness in leukemia and glioblastoma. [111],[114] Although the underlying mechanisms are unclear, differential roles for GSK3β in normal and neoplastic cells could be advantageous for cancer treatment and chemopreventive strategies that target this kinase. Animal studies conducted thus far have shown little detrimental effect of GSK3β inhibition on normal cells and vital organs. [40],[41],[43],[70]
Based on these ideas, as well as preclinical studies for the treatment of many cancer types, [24] clinical trials of GSK3β inhibitors are also being conducted for treatment of neurological diseases [115] and for enhancement of the efficacy of established chemotherapeutic agents for advanced cancers.
Acknowledgments
This work was supported in part by Grants-in-Aids for Scientific Research from the Japanese Ministry of Education, Science, Sports, Technology and Culture (to T.S., Y.M., and T.M.), and partly supported by the Extramural Collaborative Research Grant of the Cancer Research Institute, Kanazawa University.
References
| 1. | Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet 2004;363:1049-57.  [PUBMED] |
| 2. | Schneider G, Siveke JT, Eckel F, Schmid RM. Pancreatic cancer: Basic and clinical aspects. Gastroenterology 2005;128:1606-25. [PUBMED] |
| 3. | Hidalgo M. Pancreatic cancer. N Engl J Med 2010;362:1605-17. [PUBMED] |
| 4. | Fryer RA, Galustian C, Dalgleish AG. Recent advances and developments in treatment strategies against pancreatic cancer. Curr Clin Pharmacol 2009;4:102-12. [PUBMED] |
| 5. | Lockhart AC, Rothenberg ML, Berlin JD. Treatment for pancreatic cancer: Current therapy and continued progress. Gastroenterology 2005;128:1642-54. [PUBMED] |
| 6. | Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009;59:225-49. |
| 7. | Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011;61:212-36. [PUBMED] |
| 8. | Raimondi S, Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: An overview. Nat Rev Gastroenterol Hepatol 2009;6:699-708. [PUBMED] |
| 9. | Li D, Abbruzzese JL. New strategies in pancreatic cancer: Emerging epidemiologic and therapeutic concepts. Clin Cancer Res 2010;16:4313-8. |
| 10. | Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis: Aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol 2010;24:349-58. |
| 11. | Baumgart S, Ellenrieder V, Fernandez-Zapico ME. Oncogenic transcription factors: Cornerstones of inflammation-linked pancreatic carcinogenesis. Gut 2011. [Epub ahead of print] |
| 12. | Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther 2007;6:1186-97. [PUBMED] |
| 13. | Garber K. Stromal depletion goes on trial in pancreatic cancer. J Natl Cancer Inst 2010, 102:448-50. |
| 14. | Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer 2002;2:897-909. [PUBMED] |
| 15. | Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2006;20:1218-49. [PUBMED] |
| 16. | Maitra A, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Best Pract Res Clin Gastroenterol 2006;20:211-26. [PUBMED] |
| 17. | Motoo Y, Shimasaki T, Ishigaki Y, Nakajima H, Kawakami K, Minamoto T. Metabolic disorder, inflammation, and deregulated molecular pathways converging in pancreatic cancer development: Implications for new therapeutic strategies. Cancers 2011;3:446-60. |
| 18. | Giroux V, Dagorn JC, Iovanna JL. A review of kinases implicated in pancreatic cancer. Pancreatology 2009;9:738-54. [PUBMED] |
| 19. | Furukawa T. Molecular targeting therapy for pancreatic cancer: Current knowledge and perspectives from bench to bedside. J Gastroenterol 2008;43:905-11. [PUBMED] |
| 20. | Morgan MA, Parsels LA, Maybaum J, Lawrence TS. Improving gemcitabine-mediated radiosensitization using molecularly targeted therapy: A review. Clin Cancer Res 2008;14:6744-50. [PUBMED] |
| 21. | Cohen P, Goedert M. GSK3 inhibitors: Development and therapeutic potential. Nat Rev Drug Discov 2004;3:479-87. [PUBMED] |
| 22. | Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci 2004;25:471-80. [PUBMED] |
| 23. | Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem Res 2007;32:577-95. [PUBMED] |
| 24. | Miyashita K, Nakada M, Shakoori A, Ishigaki Y, Shimasaki T, Motoo Y, et al. An emerging strategy for cancer treatment targeting aberrant glycogen synthase kinase 3β. Anticancer Agents Med Chem 2009;9:1114-22. |
| 25. | Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem 1980;107:519-27. |
| 26. | Woodgett JR. cDNA cloning and properties of glycogen synthase kinase-3. Methods Enzymol 1991;200:564-77. |
| 27. | Harwood AJ. Regulation of GSK-3: A cellular multiprocessor. Cell 2001;105:821-4. |
| 28. | Doble BW, Woodgett JR. GSK-3: Tricks of the trade for a multi-tasking kinase. J Cell Sci 2003;116(Pt 7):1175-86. |
| 29. | Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 2004;29:95-102. |
| 30. | Hartmann C. A Wnt canon orchestrating osteoblastogenesis. Trends Cell Biol 2006;16:151-8. |
| 31. | Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest 2006;116:1202-9. |
| 32. | Ralston SH, de Crombrugghe B. Genetic regulation of bone mass and susceptibility to osteoporosis. Genes Dev 2006;20:2492-506. |
| 33. | Kulkarni NH, Onyia JE, Zeng Q, Tian X, Liu M, Halladay DL, et al. Orally bioavailable GSK-3α/β dual inhibitor increases markers of cellular differentiation in vitro and bone mass in vivo. J Bone Miner Res 2006;21:910-20. |
| 34. | Fuchs SY, Ougolkov AV, Spiegelman VS, Minamoto T. Oncogenic β-catenin signaling networks in colorectal cancer. Cell Cycle 2005;4:1522-39. |
| 35. | Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 2008;8:387-98. |
| 36. | Rotte A, Pasham V, Eichenmuller M, Yang W, Qadri SM, Bhandaru M, et al. Regulation of basal gastric acid secretion by the glycogen synthase kinase GSK3. J Gastroenterol 2010;45:1022-32. |
| 37. | Phukan S, Babu VS, Kannoji A, Hariharan R, Balaji VN. GSK3β: Role in therapeutic landscape and development of modulators. Br J Pharmacol 2010, 160:1-19. |
| 38. | Kalderon D. Similarities between the Hedgehog and Wnt signaling pathways. Trends Cell Biol 2002;12:523-31. |
| 39. | Manoukian AS, Woodgett JR. Role of glycogen synthase kinase-3 in cancer: Regulation by Wnts and other signaling pathways. Adv Cancer Res 2002;84:203-29. |
| 40. | Shakoori A, Ougolkov A, Yu ZW, Zhang B, Modarressi MH, Billadeau DD, et al. Deregulated GSK3β activity in colorectal cancer: Its association with tumor cell survival and proliferation. Biochem Biophys Res Commun 2005;334:1365-73. |
| 41. | Shakoori A, Mai W, Miyashita K, Yasumoto K, Takahashi Y, Ooi A, et al. Inhibition of GSK-3β activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci 2007;98:1388-93. |
| 42. | Miyashita K, Kawakami K, Nakada M, Mai W, Shakoori A, Fujisawa H, et al. Potential therapeutic effect of glycogen synthase kinase 3β inhibition against human glioblastoma. Clin Cancer Res 2009; 15:887-97. |
| 43. | Mai W, Kawakami K, Shakoori A, Kyo S, Miyashita K, Yokoi K, et al. Deregulated GSK3β sustains gastrointestinal cancer cells survival by modulating human telomerase reverse transcriptase and telomerase. Clin Cancer Res 2009;15:6810-9. |
| 44. | Shimasaki T, Ishigaki Y, Nakamura Y, Takata T, Nakaya N, Nakajima H, et al. Glycogen synthase kinase 3β inhibition sensitizes pancreatic cancer cells to gemcitabine. J Gastroenterol 2012;47:321-33. |
| 45. | Minamoto T, Kotake M, Nakada M, Shimasaki T, Motoo Y, Kawakami K. Distinct pathologic role for glycogen synthase kinase 3b in colorectal cancer progression. In: Ettarh R, editor. Colorectal Cancer Biology – From Genes to Tumor, ISBN: 978-953-51-0062-1. Rijeka, Croatia: InTech; 2012. p. 107-34. |
| 46. | Luo J. Glycogen synthase kinase 3β (GSK3β) in tumorigenesis and cancer chemotherapy. Cancer Lett 2009;273:194-200. |
| 47. | Hart AR, Kennedy H, Harvey I. Pancreatic cancer: A review of the evidence on causation. Clin Gastroenterol Hepatol 2008;6:275-82. |
| 48. | Gerdes B, Ramaswamy A, Simon B, Pietsch T, Bastian D, Kersting M, et al. Analysis of β-catenin gene mutations in pancreatic tumors. Digestion 1999;60:544-8. |
| 49. | White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology 2012;142:219-32. |
| 50. | Larsson SC, Orsini N, Wolk A. Body mass index and pancreatic cancer risk: A meta-analysis of prospective studies. Int J Cancer 2007;120:1993-8. |
| 51. | Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008;371:569-78. |
| 52. | Huxley R, Ansary-Moghaddam A, Berrington de Gonzalez A, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: A meta-analysis of 36 studies. Br J Cancer 2005;92:2076-83. |
| 53. | Luo J, Iwasaki M, Inoue M, Sasazuki S, Otani T, Ye W, et al. Body mass index, physical activity and the risk of pancreatic cancer in relation to smoking status and history of diabetes: A large-scale population-based cohort study in Japan–the JPHC study. Cancer Causes Control 2007;18:603-12. |
| 54. | Li D, Tang H, Hassan MM, Holly EA, Bracci PM, Silverman DT. Diabetes and risk of pancreatic cancer: A pooled analysis of three large case-control studies. Cancer Causes Control 2011;22:189-97. |
| 55. | Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology 2009;137:482-8. |
| 56. | Bolen S, Feldman L, Vassy J, Wilson L, Yeh HC, Marinopoulos S, et al. Systematic review: Comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Intern Med 2007;147:386-99. |
| 57. | Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, et al. Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev Res (Phila) 2010;3:1451-61. |
| 58. | McKay CJ, Glen P, McMillan DC. Chronic inflammation and pancreatic cancer. Best Pract Res Clin Gastroenterol 2008;22:65-73. |
| 59. | Greer JB, Whitcomb DC. Inflammation and pancreatic cancer: An evidence-based review. Curr Opin Pharmacol 2009;9:411-8. |
| 60. | Howes N, Lerch MM, Greenhalf W, Stocken DD, Ellis I, Simon P, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol 2004;2:252-61. |
| 61. | Rebours V, Boutron-Ruault MC, Schnee M, Ferec C, Maire F, Hammel P, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: A national exhaustive series. Am J Gastroenterol 2008;103:111-9. |
| 62. | Greer JB, Lynch HT, Brand RE. Hereditary pancreatic cancer: A clinical perspective. Best Pract Res Clin Gastroenterol 2009;23:159-70. |
| 63. | Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996;14:141-5. |
| 64. | Farrow B, Evers BM. Inflammation and the development of pancreatic cancer. Surg Oncol 2002;10:153-69. |
| 65. | Garcea G, Dennison AR, Steward WP, Berry DP. Role of inflammation in pancreatic carcinogenesis and the implications for future therapy. Pancreatology 2005;5:514-29. |
| 66. | Uomo I, Miraglia S, Pastorello M. Inflammation and pancreatic ductal adenocarcinoma: A potential scenario for novel drug targets. JOP 2010;11:199-202. |
| 67. | Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 2000;406:86-90. |
| 68. | Schwabe RF, Brenner DA. Role of glycogen synthase kinase-3 in TNF-α-induced NF-κB activation and apoptosis in hepatocytes. Am J Physiol Gastrointest Liver Physiol 2002;283:G204-11. |
| 69. | Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med 2009;361:2449-60. |
| 70. | Ougolkov AV, Fernandez-Zapico ME, Bilim VN, Smyrk TC, Chari ST, Billadeau DD. Aberrant nuclear accumulation of glycogen synthase kinase-3β in human pancreatic cancer: Association with kinase activity and tumor dedifferentiation. Clin Cancer Res 2006;12:5074-81. |
| 71. | Cao Q, Lu X, Feng YJ. Glycogen synthase kinase-3β positively regulates the proliferation of human ovarian cancer cells. Cell Res 2006;16:671-7. |
| 72. | Kotliarova S, Pastorino S, Kovell LC, Kotliarov Y, Song H, Zhang W, et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-κB, and glucose regulation. Cancer Res 2008;68:6643-51. |
| 73. | Bilim V, Ougolkov A, Yuuki K, Naito S, Kawazoe H, Muto A, et al. Glycogen synthase kinase-3: A new therapeutic target in renal cell carcinoma. Br J Cancer 2009;101:2005-14. |
| 74. | Li J, Mizukami Y, Zhang X, Jo WS, Chung DC. Oncogenic K-ras stimulates Wnt signaling in colon cancer through inhibition of GSK-3β. Gastroenterology 2005;128:1907-18. |
| 75. | Zhang B, Ougolkov A, Yamashita K, Takahashi Y, Mai M, Minamoto T. β-Catenin and ras oncogenes detect most human colorectal cancer. Clin Cancer Res 2003;9:3073-9. |
| 76. | Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998;58:1130-4. |
| 77. | Deramaudt T, Rustgi AK: Mutant KRAS in the initiation of pancreatic cancer. Biochim Biophys Acta 2005;1756:97-101. |
| 78. | Zhang JS, Koenig A, Harrison A, Ugolkov AV, Fernandez-Zapico ME, Couch FJ, et al. Mutant K-Ras increases GSK-3β gene expression via an ETS-p300 transcriptional complex in pancreatic cancer. Oncogene 2011;30:3705-15. |
| 79. | Watanabe I, Hasebe T, Sasaki S, Konishi M, Inoue K, Nakagohri T, et al. Advanced pancreatic ductal cancer: Fibrotic focus and β-catenin expression correlate with outcome. Pancreas 2003;26:326-33. |
| 80. | Tanaka Y, Kato K, Notohara K, Hojo H, Ijiri R, Miyake T, et al. Frequent β-catenin mutation and cytoplasmic/nuclear accumulation in pancreatic solid-pseudopapillary neoplasm. Cancer Res 2001;61:8401-4. |
| 81. | Makrilia N, Kollias A, Manolopoulos L, Syrigos K. Cell adhesion molecules: Role and clinical significance in cancer. Cancer Invest 2009;27:1023-37. |
| 82. | Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase-3β participates in nuclear factor κB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res 2005;65:2076-81. |
| 83. | Wilson W 3 rd, Baldwin AS. Maintenance of constitutive IκB kinase activity by glycogen synthase kinase-3α/β in pancreatic cancer. Cancer Res 2008;68:8156-63. |
| 84. | Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat Rev Cancer 2009;9:265-73. |
| 85. | Machesky LM. Lamellipodia and filopodia in metastasis and invasion. FEBS Lett 2008;582:2102-11. |
| 86. | Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol 2004;6:931-40. |
| 87. | Sun T, Rodriguez M, Kim L. Glycogen synthase kinase 3 in the world of cell migration. Dev Growth Differ 2009;51:735-42. |
| 88. | Hage B, Meinel K, Baum I, Giehl K, Menke A. Rac1 activation inhibits E-cadherin-mediated adherens junctions via binding to IQGAP1 in pancreatic carcinoma cells. Cell Commun Signal 2009;7:23. |
| 89. | Okusaka T, Kosuge T. Systemic chemotherapy for pancreatic cancer. Pancreas 2004;28:301-4. |
| 90. | Ben-Josef E, Lawrence TS. Chemoradiotherapy for unresectable pancreatic cancer. Int J Clin Oncol 2008;13:121-6. |
| 91. | Okusaka T, Ito Y, Furuse J, Yamada S, Ishii H, Shibuya K, et al. Current status of chemoradiotherapy for locally advanced pancreatic cancer in Japan. Int J Clin Oncol 2008;13:127-31. |
| 92. | Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011, 364:1817-25. |
| 93. | Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960-6. |
| 94. | Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cell 2012;21:418-29. |
| 95. | Holcomb B, Yip-Schneider M, Schmidt CM. The role of nuclear factor κB in pancreatic cancer and the clinical applications of targeted therapy. Pancreas 2008;36:225-35. |
| 96. | Mamaghani S, Patel S, Hedley DW. Glycogen synthase kinase-3 inhibition disrupts nuclear factor-κB activity in pancreatic cancer, but fails to sensitize to gemcitabine chemotherapy. BMC Cancer 2009;9:132. |
| 97. | Cano CE, Gommeaux J, Pietri S, Culcasi M, Garcia S, Seux M, et al. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res 2009;69:219-26. |
| 98. | Tomasini R, Samir AA, Vaccaro MI, Pebusque MJ, Dagorn JC, Iovanna JL, et al. Molecular and functional characterization of the stress-induced protein (SIP) gene and its two transcripts generated by alternative splicing. SIP induced by stress and promotes cell death. J Biol Chem 2001;276:44185-92. |
| 99. | Okamura S, Arakawa H, Tanaka T, Nakanishi H, Ng CC, Taya Y, et al. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol Cell 2001;8:85-94. |
| 100. | Gironella M, Seux M, Xie MJ, Cano C, Tomasini R, Gommeaux J, et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci U S A 2007;104:16170-5. |
| 101. | Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, et al. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol 2001;21:4684-99. |
| 102. | Nakahira S, Nakamori S, Tsujie M, Takahashi Y, Okami J, Yoshioka S, et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer 2007;120:1355-63. |
| 103. | Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol 2007;14:3629-37. |
| 104. | Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 2009;69:2400-7. |
| 105. | Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 2009;69:5820-8. |
| 106. | Shah AN, Gallick GE. Src, chemoresistance and epithelial to mesenchymal transition: Are they related? Anticancer Drugs 2007;18:371-5. |
| 107. | Yuan H, Mao J, Li L, Wu D. Suppression of glycogen synthase kinase activity is not sufficient for leukemia enhancer factor-1 activation. J Biol Chem 1999;274:30419-23. |
| 108. | Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, et al. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature 2005;438:873-7. |
| 109. | Takenaka K, Kise Y, Miki H. GSK3β positively regulates Hedgehog signaling through Sufu in mammalian cells. Biochem Biophys Res Commun 2007;353:501-8. |
| 110. | Surjit M, Lal SK. Glycogen synthase kinase-3 phosphorylates and regulates the stability of p27kip1 protein. Cell Cycle 2007;6:580-8. |
| 111. | Wang Z, Smith KS, Murphy M, Piloto O, Somervaille TC, Cleary ML. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 2008;455:1205-9. |
| 112. | Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 2004;10:55-63. |
| 113. | Trowbridge JJ, Xenocostas A, Moon RT, Bhatia M. Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat Med 2006;12:89-98. |
| 114. | 114 Korur S, Huber RM, Sivasankaran B, Petrich M, Morin P Jr, Hemmings BA, et al. GSK3β regulates differentiation and growth arrest in glioblastoma. PLoS One 2009;4:e7443. |
| 115. | Chico LK, Van Eldik LJ, Watterson DM. Targeting protein kinases in central nervous system disorders. Nat Rev Drug Discov 2009;8:892-909. |
Authors
Dr. Takeo Shimasaki, Medical Research Institute, Kanazawa Medical University and Hospital, Ishikawa, Japan.
Miss Ayako Kitano, Division of Translational and Clinical Oncology, Cancer Research Institute and Cancer Center, Kanazawa University and Hospital, Kanazawa, Japan.
Dr. Yoshiharu Motoo, Medical Research Institute and Department of Medical Oncology, Kanazawa Medical University and Hospital, Ishikawa, Japan.
Dr. Toshinari Minamoto, Division of Translational and Clinical Oncology, Cancer Research Institute and Cancer Center, Kanazawa University and Hospital, Kanazawa, Japan.
Figures
[Figure 1], [Figure 2]